


تکنیک ها، ویکی ژن
طراحی دارو به کمک کامپیوتر (Computer-Aided Drug Design (CADD)): انواع، موارد استفاده، مثالها و نرمافزارها

مفهوم CADD به دلیل گزینشپذیری بالاتر، کارایی، درجه تاثیر بالاتر، مدت زمان کوتاه، سمیت کم و سازگاری بهتر با پارامترهای مختلف فارماکوکینتیک (Pharmacokinetic) توجهات را به خود جلب کرده است (Makrynitsa و همکاران (2018)). تعادل مناسب شیمی دارویی (Pharmaceutical chemistry) با فعالیت بیولوژیکی مهمترین عامل در طراحی موثر دارو میباشد.
با تقاضای روزافزون داروها، انتخاب ساختارهای فعال از نظر بیولوژیکی توجه بیشتری را به خود جلب میکند. امروزه استفاده از دانش بیولوژی ساختاری و قدرت کامپیوترها جهت به کارگیری روشهای محاسباتی در شناسایی کتابخانههای فعال برای حل مشکل چاقی مولکولی، امکانپذیر شده است.
طراحی دارو به کمک کامپیوتر (CADD) چیست؟
هدف اصلی CADD غربالگری، بهینهسازی و ارزیابی فعالیت یک ترکیب در برابر هدف است. این طراحی یک رویکرد چند رشتهای را برای دانشگاههای داروسازی و شرکتهای داروسازی بزرگ برای اثربخشی بهتر و بدون عوارض جانبی یا عوارض کمتر ایجاد میکند.
پیشرفت در CADD بر اساس شباهت، شناسایی هدف، پیشبینی ساختار، محل اتصال/ حفره (Cavity) ، اعتبارسنجی، درک برهمکنش پروتئین-لیگاند (Protein-ligand interaction) به همراه غربالگری مجموعه وسیعی از ترکیبات، درک شبیهسازیهای دینامیک مولکولی بر اساس شرایط فیزیولوژیکی، محاسبه با خواص ADMET، برآورد فعالیت بیولوژیکی با استفاده از QSAR (رابطه ارزیابی کیفی مبتنی بر ساختار (Qualitative structure-based assessment relationship)) که برای کارایی و گزینشپذیری بهتر ترکیب پیشرو (Lead compound) مورد نیاز است، میباشد.
در جدول ذیل فهرست داروها بر اساس طراحی به کمک کامپیوتر آورده شده است:
| دارو | بیماری | هدف |
| اکسیمورفون (Oxymorphone) | مسکن اپیوئیدی (Opioid analgesic) | آگونیست (Agonist) گیرنده مو اپیوئیدی (mu- opioid) |
| ساکویناویر (Saquinavir) | ایدز | پروتئازهای (Protease) HIV1 و HIV 2 را مهار میکند. |
| کاپتوپریل (Captopril) | فشار خون بالا (Hypertension) | از تبدیل آنزیم مبدل آنژیوتانسین (Angiotensin) جلوگیری میکند. |
| زانامیویر (Zanamivir) | آنفولانزای A و آنفولانزای B | نورآمینیداز (Neuraminidase) را مهار میکند. |
| دورزولامید (Dorzolamide) | گلوکوم (Glaucoma) و فشار خون بالای چشم | کربنیک انیدراز (Carbonic anhydrase) را مهار میکند. |
انواع CADD
الف. طراحی دارو مبتنی بر ساختار (Structure-Based Drug Designing (SBDD))
طراحی دارو مبتنی بر ساختار چیست؟
کارآمدترین و قویترین فرآیندی که بر کل پروسه کشف دارو حاکم است، طراحی دارو مبتنی بر ساختار (SBDD) است. اطلاعات مربوط به هدف مولکولهای کوچک، اطلاعات ژنتیکی با توالیهای آنها، اطلاعات اتصال، سمیت سلولی (Cytotoxicity)، دادههای جذب، متابولیسم و دفع (ADMET) و سایر اطلاعات مهم بیولوژیکی، به عنوان کارآمدترین منابع برای تسریع فرآیند کشف دارو هستند.
این نوع طراحی به یک تکنیک محاسباتی امیدوارکننده تبدیل شده است که توسط بخش تحقیق و توسعه شرکتهای دارویی مختلف استفاده میشود. این پروسه شامل پیشبینی ساختار، تصویرسازی، مشخصهیابی محل اتصال، اتصال مولکولی و غربالگری مجازی، تصویرسازی ساختارهای پیچیده به هم متصل و آنالیز پایداری آنها، غربالگری ADMET و محاسبه انرژی اتصال آزاد با اسکریپت پایتون MM PBSA میشود.
مراحل یا فرآیند CADD مبتنی بر ساختار
- تعیین ساختار پروتئین
- مدلسازی تطبیقی
- شناسایی محل حفره در پروتئین
- تابع امتیازدهی
- امتیازدهی مبتنی بر دانش
- تابع امتیازدهی مبتنی بر نیرو
- الگوریتمهای اتصال پروتئین-لیگاند
- غربالگری مجازی
- تصویریسازی نمودار پروتئین لیگاند.
نرم افزارهای طراحی دارو مبتنی بر ساختار (SBDD)
فهرست ابزارها و برنامههای کاربردی در رویکرد SBDD در جدول ذیل آمده است:
| مرحله | ابزار مورد استفاده | توضیح کوتاه | لینکها | ||
| 1. مدلسازی هدف | SWISS-MODEL | مدلسازی هومولوژی (Homology) | SWISS-MODEL (expasy.org) | ||
| MODELER | مدلسازی هومولوژی |
|
|||
| Phyre و Phyre2 | همترازی تشخیص الگو و همچنین مدلسازی سه بعدی | PHYRE2 Protein Fold Recognition Server (ic. ac. the UK) | |||
| HHpred | تشخیص الگو، تراز و مدلسازی سه بعدی | The HHpred interactive server for protein homology detection and structure prediction | HSLS (pitt.edu) | |||
| I-TASSER | مدلسازی رشته به روش ab initio | I-TASSER server for protein structure and function prediction (zhanggroup.org) | |||
| 2. محل اتصال | CASTp | پیشبینی محل اتصال | CASTp 3.0: Computed Atlas of Surface Topography of proteins (uic.edu) | ||
| Active site prediction tool | پیشبینی سایت فعال | ACTIVE SITE PREDICTION SERVER (scfbio-iitd.res.in) | |||
| 3. اتصال مولکولی | AutoDock Vina | اتصال مولکولی و غربالگری مجازی | AutoDock Vina (scripps.edu) | ||
| Schrodinger | نرمافزار Maestro که همه همه تکنولوژیهای محاسباتی شرودینگر را شامل میشود. |
|
|||
| 4. پیشبینی ADMET | admetSAR | پیشبینی ADMET |
|
||
| QIKPROP | آنالیز ADME |
|
|||
| 5. شبیهسازی دینامیک مولکولی | AMBER | برنامههای شبیهسازی بیومولکولی را اجرا میکند. | www.gromacs.org | ||
| GROMACS | برنامه شبیهسازی دینامیک مولکولی با هدف عمومی | https://www.gromacs.org
|
|||
| MM-PBSA | g_mmpbsa | این ابزار با استفاده از روش MM-PBSA انرژی اتصال را محاسبه میکند. | ambermd.org |
محل اتصال و پیشبینی حفره:
قابل اطمینانترین و دقیقترین ناحیه در ساختار سه بعدی کمپلکس پروتئین لیگاند، آنالیز و پیشبینی ناحیهای در یک پروتئین است که لیگاند میتواند در بهترین حالت خود آن را اشغال کند. برخی ابزارهای پیشبینی حفره مانند Castro، Q-site Finder و COACH میتوانند موقعیت احتمالی محل اتصال در یک پروتئین را ارائه دهند.
این ابزارها بر اساس رویکردهای تکاملی، مبتنی بر انرژی، یا مبتنی بر ژئومتریک طراحی شدهاند. رویکرد مبتنی بر تکامل، اطلاعات مربوط به همه پروتئینهای مرتبط را میگیرد و به طور تقریبی محل اتصال مشابه را نشان میدهد. رویکردهای مبتنی بر ژئومتریک بر اساس ویژگیهایی مانند شکل، سطح آبگریز و زیرواحد سطحی باردار هستند، در حالی که رویکرد مبتنی بر انرژی از یک کاوشگر برای شناسایی مناطق اتصال مطلوب در پروتئین استفاده میکند.
نمونههایی از طراحی دارو مبتنی بر ساختار (SBDD)
فهرست منابع پایگاه داده مهم در SBDD در جدول زیر آورده شده است:
| پایگاه داده | توضیح | لینک |
| Chemspider | پایگاه داده ساختار لیگاند | ChemSpider | Search and share chemistry |
| PubChem | پایگاه داده مولکولهای کوچک همراه با فعالیت بیولوژیکی | PubChem (nih.gov) |
| DrugBank | داروهایی را که FDA برای استفاده به صورت چند منظوره تایید کرده است. | DrugBank Online | Database for Drug and Drug Target Info |
| ZINC | پایگاه دادهای که ترکیباتی را برای غربالگری مجازی و اتصال مولکولی فراهم میکند | ZINC (docking.org) |
چشمانداز آینده
ترکیب هوش مصنوعی و یادگیری ماشین با تکنیکهای مبتنی بر ساختار به عنوان ابزاری جدید با پتانسیل بسیار زیاد در کشف درمانها پدیدار شده است. شاید جامعه علمی باید بر امکانپذیری مرتبط با الگوریتمها، توابع امتیازدهی و در دسترس بودن دادههای موجود غلبه کند. این چارچوب اساسی آزمایشهای بالینی را تسریع و عدم موفقیت نامزدهای دارویی امیدوارکننده را محدود میکند. از این پس، SBDD میتواند به عنوان یک تکنیک جدید، قابل اعتماد و کارآمد برای طراحی دارو ظاهر شود.
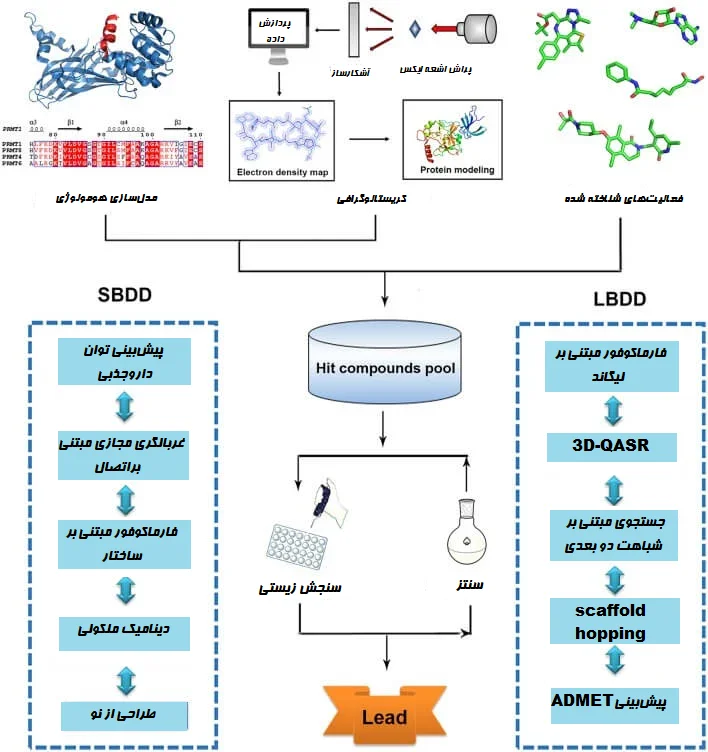
ب. طراحی دارو مبتنی بر لیگاند (Ligand-based drug designing (LBDD))
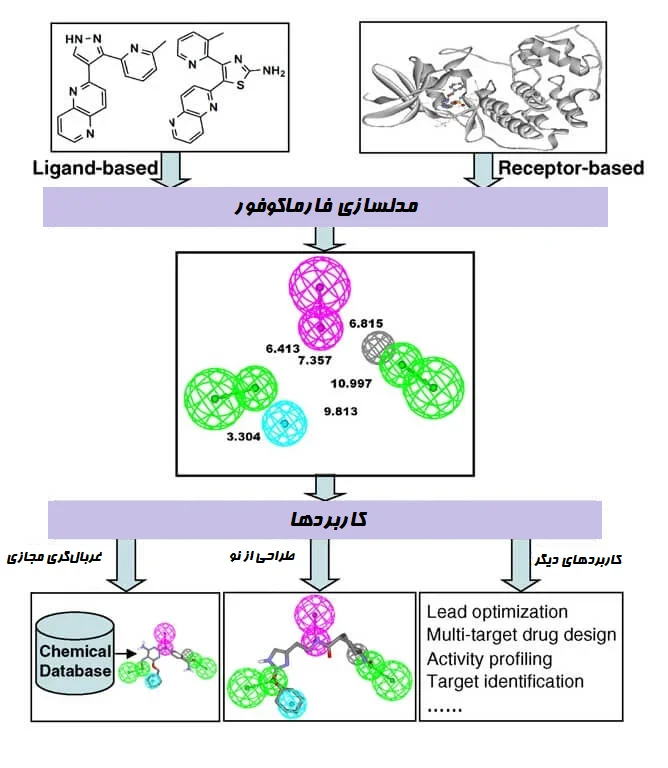
طراحی دارو مبتنی بر لیگاند (LBDD) چیست؟
عدم وجود ساختار ماکرومولکولی سه بعدی منجر به شناسایی بسیار سیستماتیک هدف با استفاده از ابزارهای محاسباتی پیشرفته میشود. محققان داروسازی در حال حاضر با شرایط سختتر و چالش بزرگتری مواجه شدهاند.
فرضیه غربالگری با توان عملیاتی بالا (high-throughput screening (HTS)) با استراتژیهای در حال ظهور بهتر تقویت شده است. روش غیرمستقیم طراحی دارو یا طراحی دارو مبتنی بر لیگاند به طور گستردهای از این چالش سختتر پشتیبانی میکند. این طراحی را میتوان به سه روش مختلف زیر انجام داد:
- رویکرد یادگیری ماشینی
- روشهای جستجوی شباهت
- طراحی مدلهای فارماکوفور
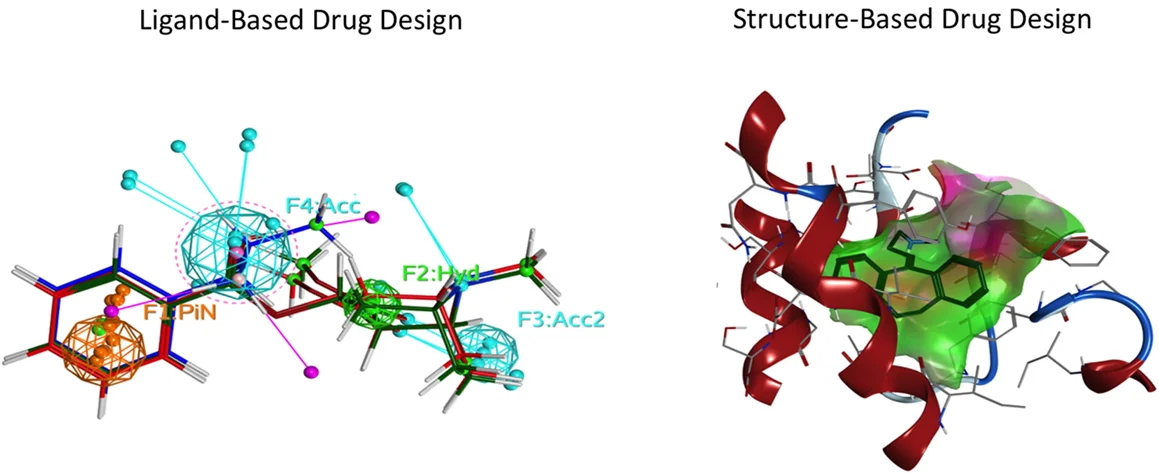
نقشهبرداری مدلهای فارماکوفور
این رویکرد در بهینهسازی پیشرو (Lead optimization) در شیمی پزشکی و محاسباتی استفاده میشود. این رویکرد بر اساس استفاده از همترازسازی ساختاری و فعالیتهای پیشبینی و جستجوی پایگاه داده سه بعدی ساخته شده است. در اینجا تعدادی از مجموعه دادههای شناخته شده یا ناشناخته برای توسعه ساختار مولکولی همراه با فعالیت آن نسبت به هر هدف، استفاده میشود.
این کار را میتوان در زمینههایی مانند تعیین مشخصههای ADMET، تخمین عوارض جانبی و تعیین پیشبینیهای غیرصحیح انجام داد. با توجه به دانش فارماکوفور، مطالعات شبیهسازی با غربالگری مجازی بهبود یافته است.
مراحل اساسی مورد استفاده در مدلسازی فارماکوفور
- تعیین مجموعهای متنوع از ساختارها با فعالیتهای تجربی به درستی تعریف شده
- تولید ترکیبات
- بکارگیری مدلهای فارماکوفور سه بعدی در مجموعه آموزشی از ترکیبات در سه فاز
- فاز سازنده: مدلهای فارماکوفور بر روی مولکولهای فعال مشترک ساخته میشوند.
- فاز کاهش: بر خلاف مدلهای فارماکوفور، کاهش میدهد.
- فاز بهینه سازی: با بهینه سازی یک مدل، فرضیه ایجاد میشود.
- ارزیابی کیفیت: انتخاب فارماکوفور بر اساس تابع هزینه انجام میشود.
- اعتبار سنجی
- آزمایش آرایش تصادفی فیشر (Fisher) بر روی مجموعهای از ترکیبات معتبر انجام میشود.
- در نهایت، نقشهبرداری بر روی مجموعهای آزمایشی از ترکیبات انجام میشود.
ابزارهای مورد استفاده برای مدلسازی و کاربردهای فارماکوفور
| ردیف | ابزار | توضیح | لینک |
| 1 | Pharmer | این ابزار از ترکیباتی برای غربالگری مجازی استفاده میکند. | Welcome to ZINCPharmer (pitt.edu) |
| 2 | Pharmapper | این ابزار شامل دانش بیش از 7000 مدل فارماکوفور مبتنی بر هدف است و بهترین تطابق را برای ورودی به عنوان لیگاند در برابر مدلهای مبتنی بر فارماکوفور فراهم میکند. | Welcome to our lab (lab-request.cn) |
| 3 | pharmacist | این ابزار از مجموعهای از لیگاندها در غیاب هدف جستجو را انجام میدهد. | BioInfo3D Group (tau.ac.il) |
| 4 | Pharmit tool | این ابزار بر بررسی دارو در فارماکوکینتیک (Pharmacokinetics) متکی است | pharmit: interactive exploration of chemical space (pitt.edu) |
| 5 | Discovery Studio | ابزاری که برای درمان و نظارت بر دارو استفاده میشود. | Free Download: BIOVIA Discovery Studio Visualizer – Dassault Systèmes (3ds.com) |
| 6 | Ligandscout | این ابزار فارماکوپورهای سه بعدی را از دادههای ساختاری به عنوان مجموعه آموزشی/ آزمایشی از مولکولها، مدلسازی میکند. | Linux |
| 7 | Phase | این ابزار طراحی دارویی مبتنی بر هم لیگاند و هم ساختار را انجام میدهد. | Schrödinger | Schrödinger is the scientific leader in developing state-of-the-art chemical simulation software for use in pharmaceutical, biotechnology, and materials research. (schrodinger.com) |
کشف مولکول پیشرو یا رهبر (Lead molecule)
ترکیب شیمیایی که دارای خواص بیولوژیکی یا دارویی با ویژگیهای درمانی است را میتوان مولکول پیشرو نامید. این خواص شامل پارامترهایی مانند گزینشپذیری خوب، کارایی، درجه تاثیر بهتر همراه با ابزارهای کینتیک دارویی (Pharma kinetic) میباشد. با این وجود، بیشتر ترکیبات پیشرو تحت اصلاحات ساختاری قرار میگیرند تا خواص بیولوژیکی خاص خود را برآورده کنند (این فرایند به اصطلاح بهینهسازی پیشرو نام دارد). با این حال، ترکیب پیشرو مورد بررسی باید پنج ویژگی را برآورده کند تا به عنوان یک مولکول دارویی زیستفعال (Bioactive) عمل کند.
این ویژگیها شامل قدرت، مدت زمان، ایمنی، در دسترس بودن و مقبولیت دارویی هستند، که در آن قدرت شامل توانایی هر مولکول برای نشان دادن خواص دارویی مطلوب در مقادیر کمتر است. انتقال ترکیبات دارویی با موانع متعدد برای رسیدن به محل مورد نظر، فراهمیزیستی (Bioavailability) نامیده میشود.
مفهوم مدت زمان با زمان ورود مولکولهای دارو همراه با پاسخ دارویی آنها مشخص میشود. قوانین ایمنی شامل پارامترهای سمیتی است که بدون عوارض جانبی بر ارگانیسمهای هدف بوده یا عوارض جانبی کمتری بر آنها از خود نشان میدهد. از این پس همه فاکتورهای ذکر شده در بالا به طور مساوی در بهینهسازی پیشروی مولکول دارو نقش دارند. هدف از کشف سریع داروها شامل شناسایی و اعتبارسنجی هدف است. پس از فرآیند اعتبارسنجی هدف، شناسایی مراحل کشف Hit to Lead باید در یک فرآیند یافتن داروی جدید توسعه یابد.
به هر حال پارامترهای فیزیکی و بیوشیمیایی برای تصمیمگیری در مورد تغییر در ویژگی ساختاری ترکیبات برای سنتز یک مولکول پیشروی موثر در توسعه دارو استفاده شده است. تعدادی ملکول پیشرو طبیعی با فعالیت بیولوژیکی در برابر هدف خاص خود که منجر به اصلاح شیمیایی میشوند، در پایگاههای اطلاعاتی و ادبیاتی مختلف در دسترس قرار گرفتهاند. آنها به 3 دسته طبیعی، سنتتیک (Synthetic) و نیمه سنتتیک تقسیم میشوند.
بهینهسازی پیشرو و استراتژیهای آن
هر ترکیب پیشرو را میتوان از نظر ساختاری اصلاح کرد و خواص فیزیکوشیمیایی، ترمودینامیکی، پارامترهای فارماکوکینتیک و سمیت آن را حفظ کرد (Jogensen, 2009). طبق قانون لیپینسکی (Lipinski)، دارو باید از چند ویژگی مانند داشتن کمتر از 5 پیوند هیدروژنی و 10 پیوند پذیرنده تبعیت کند. جرم مولکولی باید کمتر از 500 دالتون باشد و ضریب تقسیم (Partition coefficient یا p-log) آن نباید بیشتر از 5 باشد (Lipinsk و همکاران (2003)).
بهینهسازی پیشرو تنها به پارامترهای اتصالی بستگی دارد که اطلاعاتی در مورد اثربخشی دارو و همچنین فرآیند متابولیک موجود در ارگانیسمها را نشان میدهند. شاخه شیمی سنتتیک آلی شامل سادهسازی ساختار، اصلاح، پارامترهای دینامیکی و کینتیکی، تبدیل گروه عاملی و انتخابی بودن یا نیروی پیوند میباشد. پارامترهای دیگر مانند کینتیکی شامل ترمودینامیک، فارماکودینامیک همراه با بهبود بیشتر در خواص ADMET نیز باید در نظر گرفته شوند.
در زیر لیستی از ترکیبات فعال طبیعی و کاربردهای آنها آورده شده است:
| ردیف | ترکیبات طبیعی | آنالوگهای فعال | کاربرد |
| 1 | مورفولین (Morpholine) | بوتورفانول (Butorphanol)، پنتازوسین (Pentazocine)، متادون (Methadone) | آگونیست و آنتاگونیست (Antagonist) گیرنده مواد اپیوئیدی را هدف قرار میدهد. |
| 2 | هالیکندرین B (Halichondrin B) | اریبولین (Eribulin)، مزیلات (Mesylate) | فعالیت ضد تومور |
| 3 | میریوسین (Myriocin) | فینگولیمود (Fingolimod) | عامل سرکوبکننده سیستم ایمنی |
| 4 | تریکوستاتین A (Trichostatin A) | ورینوستات (Vorinstat) | مهارکننده هیستون داستیلاز (Histone deacetylase) |
| 5 | شیسندین C (Schisandin C) | Bicyclol | فعالیت ضد هپاتیت B |
| 6 | آسپرلیسین (Asperlicin) | دوازپید (Devazepide) | آنتاگونیست گیرنده کولهسیستوکینین (Cholecystokinin) |
روندهای اخیر در طراحی دارو
تغییر کاربری داروها (بازکاربرد دارویی (Drug repositioning)، دستهبندی مجدد، یا به کارگیری مجدد) روشی برای شناسایی تمام کاربردهای بالقوهای است که فراتر از موارد مصرف پزشکی واقعی یا داروهای تحقیقاتی هستند (Ashburn و Thor (2004)). این مفهوم را میتوان به عنوان دستهبندی مجدد دارویی (Drug reprofiling) به معنی کشف موارد مصرف جدید از هر یک از ترکیبات تایید شده / ناموفق / رها شده برای استفاده در بیماریهای مختلف، تعریف کرد.
نتیجه
CADD به عنوان یک رویکرد چند رشتهای جهت توسعه داروها در دوران مدرن عمل میکند. یکی از چالشهای اصلی، تولید دارویی است که به خوبی برای بیماران مناسب باشد و اثربخشی بهتری داشته باشد.
رویکردهای CADD دقیقتر و کارآمدتر شدهاند. ایجاد یک بهبود قابل توجه در جستجوی ترکیبات با پلتفرم جستجوی مشابهت، شناسایی هدف، پیشبینی ساختار، پیشبینی/ اعتبارسنجی فعالیت محل اتصال، دانش اثر متقابل پروتئین-پروتئین در شرایط فیزیولوژیکی، محاسبه خواص ADMET، تخمین فعالیت بیولوژیکی با استفاده از ابزارهایی مانند QSAR، تغییرات لازم را برای رسیدن به یک ترکیب پیشرو جهت بهبود کارایی و همچنین گزینشپذیری هدایت میکند.
کشف یک داروی مدرن موجود مستلزم داشتن دانش ژنومیک، بیوانفورماتیک، شیمی محاسباتی و شیمی ترکیبی برای غربالگری مجازی، HTS و لیگاندهای Lenovo است. با این حال، مفهوم LBDD بیشتر بر طراحی دارویی متکی است که یک مولکول دارویی ساده را به یک مولکول سه بعدی از طریق روشهای آزمون و خطا که فعالیت بیولوژیکی کلی ترکیب را بهبود میبخشد، توسعه میدهد. در این مقاله به چند نرم افزار و اصول طراحی دارو مبتنی بر لیگاند پرداخته شد.
کاربردهای طراحی دارو به کمک کامپیوتر (CADD)
CADD توجه زیادی را به سه عامل اصلی جلب میکند:
- غربالگری مجموعه بزرگی از مولکولها در ساختار هدف، که به صورت محاسباتی و همچنین تجربی ارزیابی میشود،
- هدایت بهینهسازی ترکیبات پیشرو بسته به میل ترکیبی، پارامترهای فارماکوکینتیک و همچنین سمیت آنها
- طراحی ترکیبات جدید بسته به ساختارشان برای بهبود عملکرد آنها به عنوان یک ترکیب دارویی.
به کارگیری CADD در مدلسازی دارو، به شیمی ترکیبی و بیوانفورماتیک که به مسائل عمده از جمله هزینه و مدت زمان میپردازند، رسیدگی میکند.
محدودیتهای طراحی دارو به کمک کامپیوتر (CADD)

اگرچه رویکرد SBDD به عنوان یک رویکرد پیشگام در طراحی دارو ثابت شده است، اما باید از چالشهایی عبور کند که جامعه باید به آنها نگاه کند. این رویکردهای پیشرو شامل روشهای غربالگری، ترکیبات شیمیایی، بهبود دادهها، کمیت و کیفیت ابزارها و پایگاههای داده مختلف، اصلاح ساختارهای دارویی چند هدفی، الگوریتمهای پیشبینی سمیت، ادغام رویکرد برای کارایی و سازگاری بهتر میباشند.
پارامترهای دیگر با بیشترین اهمیت، برهمکنشهای الکترواستاتیکی و محاسبات آنتروپی هستند که به طور کامل نادیده گرفته شدهاند. مهمتر از همه، هیچ نرمافزار یا پکیجی وجود ندارد که برای اهداف و لیگاندهای خاص از جمله مولکول آب به خوبی کار کند و تأیید احتمالی برای هدف و سایر نوآوریها هنوز باید بررسی شوند.
همچنین بخوانید:
- دوره مهارت آموزی طراحی دارو
- هومولوژی مدلینگ با نرم افزار MODELLER
- داروهای بیوسیمیلار (Biosimilar)
- تکنیک طراحی دارو یا Drug delivery
- هوش مصنوعی در طراحی دارو
مترجم: صادق حسینیکیا