


مقدمهای بر نوروبلاستوما
سرطان زمانی شروع میشود که سلولهای بدن شروع به رشد غیر قابل کنترل میکنند و سلولهای طبیعی را از بین میبرند. سلولها تقریباً در هر قسمت از بدن میتوانند به سرطان تبدیل شوند و سپس به سایر قسمتهای بدن گسترش یابند.
نوروبلاستوما (Neuroblastoma) سرطانی است که در برخی از اشکال اولیه سلولهای عصبی شروع میشود که اغلب در جنین یا نوزاد یافت میشود. (اصطلاح نورون به اعصاب اشاره دارد، در حالی که بلاستوما به سرطانی اشاره دارد که در سلولهای نابالغ یا در حال رشد شروع میشود). این نوع سرطان بیشتر در نوزادان و کودکان خردسال رخ میدهد.
انواع سرطانهایی که در کودکان ایجاد میشود اغلب با انواعی که در بزرگسالان ایجاد میشوند، متفاوت است.
برای درک نوروبلاستوما، شناخت سیستم عصبی سمپاتیک – جایی که این تومورها در آن شروع میشوند – به شما کمک میکند.
سیستم عصبی سمپاتیک (sympathetic nervous system)
مغز، نخاع و اعصابی که از آنها به تمام قسمتهای بدن میرسند، همگی بخشی از سیستم عصبی هستند. سیستم عصبی برای تفکر، احساس و حرکت و از جمله چیزهای دیگر مورد نیاز است.
بخشی از سیستم عصبی همچنین عملکردهای بدن را که به ندرت از آنها آگاه هستیم، مانند ضربان قلب، تنفس، فشار خون، هضم و سایر عملکردها را کنترل میکند. این قسمت از سیستم عصبی به عنوان سیستم عصبی خودمختار (autonomic nervous system) شناخته میشود.
سیستم عصبی سمپاتیک بخشی از سیستم عصبی خود مختار است. این سیستم شامل:
- رشتههای عصبی که در امتداد دو طرف نخاع قرار دارند.
- خوشههایی از سلولهای عصبی به نام گانگلیا یا ganglia (جمع گانگلیون) در نقاط خاصی در طول مسیر رشتههای عصبی.
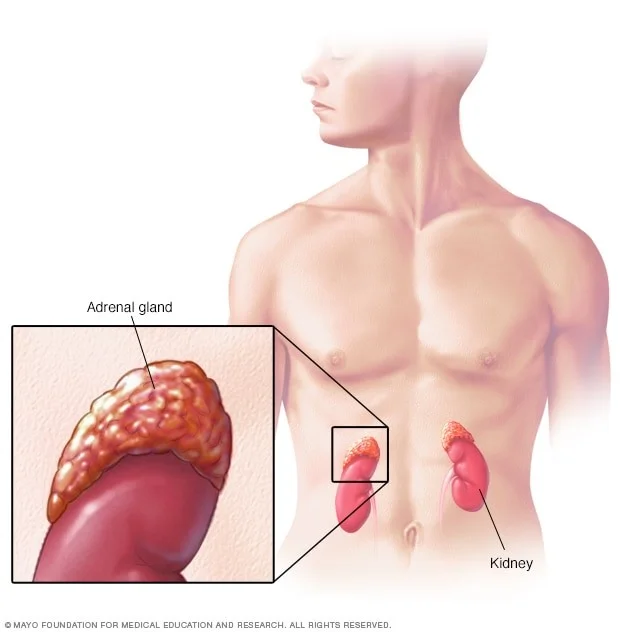
- سلولهای عصبی مانند که در بصل النخاع یا medulla (مرکز) غدد فوق کلیوی (adrenal glands) یافت میشوند. آدرنالها (adrenal glands) غدد کوچکی هستند که در بالای هر کلیه قرار دارند. این غدد هورمونهایی (مانند آدرنالین [اپی نفرین]) میسازند که به کنترل ضربان قلب، فشار خون، قند خون و نحوه واکنش بدن به استرس کمک میکند.
سلولهای اصلی تشکیل دهنده سیستم عصبی، سلولهای عصبی (nerve cells) یا نورون (neurons) نامیده میشوند. این سلولها با ترشح مقادیر کمی از مواد شیمیایی (هورمون) با انواع دیگر سلولهای بدن تعامل دارند. این مهم است زیرا سلولهای نوروبلاستوما اغلب مواد شیمیایی خاصی را آزاد میکنند که میتوانند علائم ایجاد کنند
نوروبلاستوم (Neuroblastomas)
نوروبلاستومها سرطانهایی هستند که در سلولهای عصبی اولیه (به نام نوروبلاستها یا neuroblasts) سیستم عصبی سمپاتیک شروع میشوند، بنابراین میتوانند در هر نقطه از این سیستم یافت شوند.
- اکثر نوروبلاستومها از شکم شروع میشوند یا در غده فوق کلیوی یا در گره های عصب سمپاتیک.
- بیشتر بقیه موارد در گانگلیونهای سمپاتیک نزدیک ستون فقرات در قفسه سینه یا گردن یا در لگن شروع میشوند.
- به ندرت، یک نوروبلاستوما تا زمانی که مشخص شود آن قدر گسترده شده است که پزشکان نمیتوانند دقیقاً بگویند که از کجا شروع شده است.
برخی از نوروبلاستومها به سرعت رشد میکنند و گسترش مییابند، در حالی که برخی دیگر به کندی رشد میکنند. گاهی اوقات، در کودکان بسیار کوچک، سلولهای سرطانی بدون دلیل میمیرند و تومور خود به خود از بین میرود. در موارد دیگر، سلولها گاهی به خودی خود به سلولهای گانگلیونی طبیعی بالغ میشوند و تقسیم نمیشوند (که تومور را تبدیل به گانگلیونورومای خوش خیم میکند – به مباحث زیر مراجعه کنید).
سایر تومورهای سیستم عصبی خودمختار در کودکان
همه تومورهای سیستم عصبی خود مختار دوران کودکی بدخیم (سرطانی) نیستند. برخی از تومورها خوش خیم (غیر سرطانی) هستند و برخی میتوانند هم سلولهای خوش خیم و هم سلولهای سرطانی در یک تومور داشته باشند.
- گانگلیونوروما (Ganglioneuroma) یک تومور خوش خیم (غیر سرطانی) است که از سلولهای گانگلیونی و غلاف عصبی بالغ تشکیل شده است.
- Ganglioneuroblastoma توموری است که دارای دو قسمت بدخیم و خوش خیم است. این شامل نوروبلاستها (سلولهای عصبی نابالغ) است که میتوانند به طور غیر طبیعی رشد کنند و مانند نوروبلاستوما گسترش یابند و همچنین مناطقی از بافت بالغ تر که شبیه گانگلیونوروما هستند.
اگر تصور شود که کودکی یکی از این تومورها را دارد، معمولاً با جراحی برداشته شده و با میکروسکوپ به دقت بررسی میشود تا ببینند آیا مناطقی از سلولهای سرطانی دارد (که آن را به گانگلیونوروبلاستوما تبدیل میکند). اگر تشخیص نهایی گانگلیونوروما باشد، به درمان دیگری نیاز نیست. اگر مشخص شود که بیماری گانگلیونوروبلاستوما است، مانند نوروبلاستوما درمان میشود.
آمار کلیدی در مورد نوروبلاستوما
نوروبلاستوما شایع ترین سرطان در نوزادان (کوچکتر از 1 سال) است. هر ساله حدود 700 تا 800 مورد جدید نوروبلاستوما در ایالات متحده وجود دارد. این عدد برای سالیان متمادی تقریباً ثابت مانده است.
میانگین سنی کودکان در هنگام تشخیص حدود 1 تا 2 سال است. به ندرت، نوروبلاستوم با سونوگرافی حتی قبل از تولد تشخیص داده میشود. حدود 9 مورد از هر 10 نوروبلاستوم در سن 5 سالگی تشخیص داده میشود. در افراد بالای 10 سال نادر است.
آمارهای مربوط به بقا در مبحث نرخ بقای نوروبلاستوما بر اساس گروههای خطر مورد بحث قرار گرفته است.
علائم و نشانههای نوروبلاستوما
نوروبلاستوما میتواند در نقاط مختلف بدن شروع شود. سلولهای نوروبلاستوما همچنین گاهی اوقات میتوانند مواد شیمیایی به نام هورمونها را آزاد کنند که میتوانند سایر قسمتهای بدن را تحت تأثیر قرار دهند. به همین دلیل، نوروبلاستوم میتواند علائم و نشانههای مختلفی ایجاد کند.
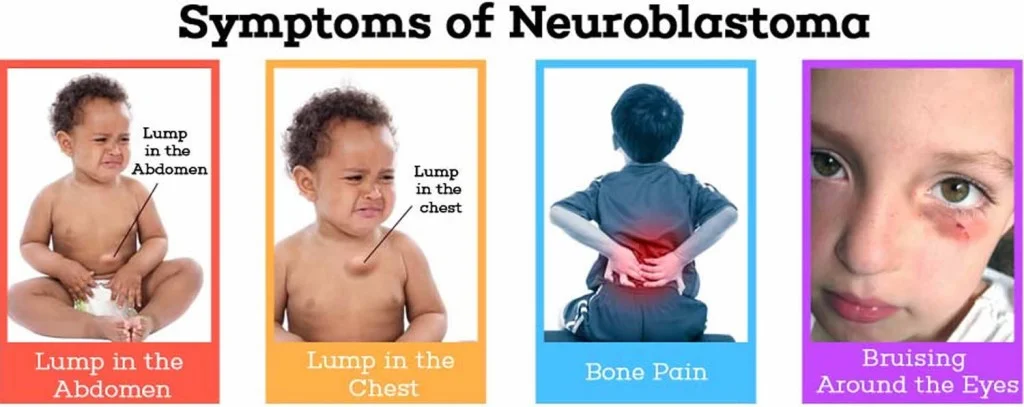
برخی از علائم رایج تر میتواند شامل موارد زیر باشد:
- توده یا تورم در شکم کودک که به نظر میرسد، درد ندارد.
- تورم در پاها یا در قسمت بالای سینه، گردن و صورت
- مشکلات تنفس یا بلع
- کاهش وزن
- نخوردن غذا یا شکایت از احساس سیری
- مشکلاتی در اجابت مزاج یا ادرار کردن
- درد در استخوانها
- تودهها یا برجستگیهای زیر پوست که ممکن است آبی به نظر برسند.
- افتادگی پلک و مردمک کوچک (ناحیه سیاه در مرکز چشم) در یک چشم
- مشکل در لمس یا حرکت دادن قسمتهایی از بدن
- چشمهایی که به نظر میرسد برآمدگی و یا کبودی در اطرافشان دارند
بسته به اینکه تومور کجاست، چقدر بزرگ است، چقدر گسترش یافته است و اینکه تومور هورمون تولید میکند، علائم و نشانهها ممکن است متفاوت باشد.
بسیاری از این علائم و نشانهها به احتمال زیاد ناشی از چیزی غیر از نوروبلاستوم است. با این حال، اگر کودک شما هر یک از این علائم را داشت، با پزشک خود مشورت کنید تا بتوان علت را پیدا کرده و در صورت نیاز آن را درمان کرد.
نشانهها یا علائم ناشی از تومور اصلی
تومورهای شکم (belly) یا لگن (pelvis): یکی از شایع ترین علائم نوروبلاستوم، وجود توده یا تورم بزرگ در شکم کودک است. ممکن است کودک تمایلی به خوردن نداشته باشد (که میتواند منجر به کاهش وزن شود). اگر کودک به اندازه کافی بزرگ شده باشد، ممکن است از احساس سیری یا درد شکم شکایت کند. اما خود توده معمولاً هنگام لمس دردناک نیست.
گاهی اوقات، تومور در شکم یا لگن میتواند سایر قسمتهای بدن را تحت تاثیر قرار دهد. به عنوان مثال، تومورهایی که به خون و عروق لنفاوی شکم یا لگن فشار میآورند یا به داخل آن رشد میکنند، میتوانند مانع از بازگشت مایعات به قلب شوند. این گاهی اوقات میتواند منجر به تورم در پاها و در پسران، کیسه بیضه شود.
در برخی موارد، فشار ناشی از یک تومور در حال رشد میتواند بر مثانه یا روده کودک تأثیر بگذارد که میتواند باعث ایجاد مشکل در ادرار کردن یا اجابت مزاج شود.
تومور در قفسه سینه یا گردن: تومورهای گردن اغلب به صورت یک توده سخت و بدون درد دیده یا احساس میشوند. اگر تومور در قفسه سینه باشد، ممکن است به ورید اجوف فوقانی یا superior vena cava (ورید بزرگ در قفسه سینه که خون را از سر و گردن به قلب باز میگرداند) فشار بیاورد. این میتواند باعث تورم در صورت، گردن، بازوها و بالای سینه شود (گاهی اوقات با رنگ پوست مایل به قرمز). همچنین اگر بر مغز تأثیر بگذارد میتواند باعث سردرد، سرگیجه و تغییر در هوشیاری شود.
تومورهای قفسه سینه یا گردن ممکن است روی گلو یا نای نیز فشار بیاورند که میتواند باعث سرفه و مشکل در تنفس یا بلع شود.
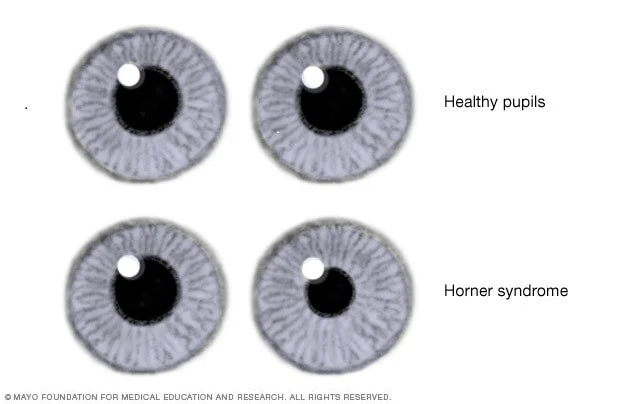
نوروبلاستومهایی که بر روی اعصاب خاصی در قفسه سینه یا گردن فشار میآورند، گاهی اوقات میتوانند علائم دیگری مانند افتادگی پلک و مردمک کوچک (ناحیه سیاه در مرکز چشم) ایجاد کنند که بخشی از بیماری شناخته شده به عنوان سندرم هورنر (Horner syndrome) است.
فشار روی سایر اعصاب نزدیک ستون فقرات ممکن است بر توانایی کودک در احساس یا حرکت بازوها یا پاهای خود تأثیر بگذارد.
نشانهها یا علائم ناشی از سرطان به سایر قسمتهای بدن گسترش مییابد
بسیاری از نوروبلاستوماها تا زمان پیدا شدن به غدد لنفاوی یا سایر قسمتهای بدن گسترش یافته اند.
انتشار به غدد لنفاوی (lymph nodes): غدد لنفاوی مجموعهای به اندازه یک لوبیا از سلولهای ایمنی هستند که در سراسر بدن یافت میشوند. سرطانی که به غدد لنفاوی گسترش یافته است میتواند باعث تورم آنها شود. این گرهها گاهی اوقات میتوانند به صورت تودههایی در زیر پوست، به خصوص در گردن، بالای استخوان ترقوه، زیر بازو یا در کشاله ران احساس شوند. بزرگ شدن غدد لنفاوی در کودکان بسیار بیشتر از سرطان نشانه عفونت است اما باید توسط پزشک بررسی شود.
گسترش به استخوانها: نوروبلاستوما اغلب به استخوانها سرایت میکند. کودکی که میتواند صحبت کند ممکن است از درد استخوان شکایت کند. درد ممکن است به قدری شدید باشد که کودک لنگ بزند یا از راه رفتن امتناع کند. اگر به استخوانهای ستون فقرات گسترش یابد، تومورها میتوانند بر روی نخاع فشار بیاورند و باعث ضعف، بی حسی یا فلج در بازوها یا پاها شوند.
انتشار به استخوانهای اطراف چشم شایع است و میتواند منجر به کبودی در اطراف چشم شده یا باعث شود کره چشم کمی بیرون بیاید. سرطان ممکن است به سایر استخوانهای جمجمه نیز سرایت کند و باعث ایجاد برجستگی در زیر پوست سر شود.
گسترش به مغز استخوان (bone marrow): اگر سرطان به مغز استخوان (قسمت داخلی برخی از استخوانها، جایی که سلولهای خونی جدید ساخته میشود) گسترش یابد، ممکن است کودک گلبولهای قرمز، گلبولهای سفید یا پلاکتهای خون کافی نداشته باشد. این کمبود سلولهای خونی میتواند منجر به خستگی، تحریکپذیری، ضعف، عفونتهای مکرر و کبودی یا خونریزی بیش از حد ناشی از بریدگیها یا خراشهای کوچک شود.
مشکلات خونریزی: به ندرت، تومورهای بزرگ میتوانند شروع به تجزیه شدن کنند که منجر به از دست دادن فاکتورهای لخته شدن خون میشود. این میتواند منجر به خطر بالای خونریزی جدی شود که به عنوان یک انعقاد مصرفی (consumption coagulopathy) شناخته میشود و میتواند تهدید کننده زندگی باشد.
مرحله S4 (MS) نوروبلاستوم: یک نوع گسترده خاص از نوروبلاستوم (معروف به مرحله S4 یا مرحله MS) گاهی اوقات، معمولا در چند ماه اول زندگی رخ میدهد. در این شکل خاص، نوروبلاستوما به کبد، پوست و یا مغز استخوان (در مقادیر کم) گسترش یافته است. برآمدگیهای آبی یا بنفش که شبیه زغال اختههای کوچک هستند ممکن است نشانه انتشار به پوست باشد. کبد میتواند بسیار بزرگ شود و به عنوان یک توده در سمت راست شکم احساس شود.
گاهی اوقات کبد میتواند به اندازه ای بزرگ شود که به ریهها فشار بیاورد که میتواند نفس کشیدن را برای کودک سخت کند. در حالی که مرحله S4 نوروبلاستوم در حال حاضر گسترش یافته است اما بسیار قابل درمان است و اغلب به خودی خود کوچک میشود یا از بین میرود. تقریباً همه کودکان مبتلا به این نوع نوروبلاستوما قابل درمان هستند.
نشانهها یا علائم ناشی از هورمونهای تومور
سلولهای نوروبلاستوما گاهی اوقات هورمونهایی ترشح میکنند که میتوانند باعث ایجاد مشکلاتی در بافتها و اندامهای سایر قسمتهای بدن شوند، حتی اگر سرطان به آن بافتها یا اندامها سرایت نکرده باشد. به این مشکلات سندرمهای پارانئوپلاستیک (paraneoplastic syndromes) میگویند.
علائم و نشانههای سندرم پارانئوپلاستیک میتواند شامل موارد زیر باشد:
- اسهال آبکی مداوم
- تب
- فشار خون بالا (باعث تحریک پذیری)
- ضربان قلب سریع
- قرمزی (گرگرفتگی) پوست
- تعریق
مجموعهای از علائم غیر معمول، سندرم اپسوکلونوس-میوکلونوس-آتاکسی (opsoclonus-myoclonus-ataxia syndrome) یا آپسوکلونوس میوکلونوس آتاکسی (OMA یا opsoclonus myoclonus ataxia) نامیده میشود.

تصور میشود که این ناشی از حمله سیستم ایمنی بدن به بافت عصبی طبیعی است. کودک مبتلا به این سندرم به طور معمول دارای حرکات نامنظم و سریع چشم (opsoclonus) و اسپاسم عضلات مانند انقباض (میوکلونوس یا myoclonus) است و هنگام ایستادن یا راه رفتن ناهماهنگ به نظر میرسد (آتاکسی یا ataxia).
آنها همچنین ممکن است در صحبت کردن مشکل داشته باشند. کودکانی که به این سندرم مبتلا هستند، در مورد خود نوروبلاستوما، دیدگاه بهتری دارند، اگرچه برخی از کودکان ممکن است مشکلات طولانی مدت سیستم عصبی داشته باشند، حتی پس از درمان نوروبلاستوم.
چه چیزی باعث نوروبلاستوما میشود؟
در حالی که چند عامل خطرزا شناخته شده برای نوروبلاستوم وجود دارد، علت اکثر نوروبلاستومها شناخته شده نیست. با این حال، محققان تفاوتهای مهمی را بین سلولهای نوروبلاستوم و نوروبلاستهای طبیعی (اشکال اولیه سلولهای عصبی) که از آنها رشد میکنند، یافته اند.
چگونه سلولهای طبیعی به سلولهای نوروبلاستوما تبدیل میشوند؟
سلولهای عصبی و سلولهای مدولا (مرکز) غده فوق کلیوی به عنوان نوروبلاست در جنین در حال رشد شروع میشوند. اغلب نوروبلاستها رشد کرده و به سلولهای بالغ تبدیل میشوند. نوروبلاستومها زمانی رخ میدهند که نوروبلاستهای طبیعی جنین به سلولهای بالغ تبدیل نمیشوند اما در عوض به رشد و تقسیم خود ادامه میدهند.
برخی از نوروبلاستها ممکن است تا زمان تولد نوزاد به طور کامل بالغ نشده باشند. اکثر اینها در نهایت در طول زمان به سلولهای عصبی طبیعی بالغ میشوند یا به سادگی از بین میروند و نوروبلاستوم تشکیل نمیدهند.
اما گاهی اوقات نوروبلاستهای باقی مانده در نوزادان بسیار جوان به رشد خود ادامه داده و تومور را تشکیل میدهند. بسیاری از این تومورها همچنان در نهایت به بافت عصبی بالغ میشوند یا خود به خود از بین میروند اما با بزرگتر شدن کودکان، احتمال بلوغ این سلولها کاهش مییابد و به سرطان تبدیل میشوند.
چرا برخی از نوروبلاستها بالغ نمیشوند؟
دلیل بالغ نشدن برخی نوروبلاستها این است که تغییراتی در DNA درون سلولها دارند. DNA ماده شیمیایی موجود در هر یک از سلولهای ما است که ژنهای ما را میسازد و عملکرد سلولهای ما را کنترل میکند. DNA سلولهای ما در ساختارهای طویل و رشته مانندی به نام کروموزوم (chromosomes) مرتب شده اند.
برخی از ژنها به طور معمول زمان رشد سلولهای ما، تقسیم به سلولهای جدید و مرگ را کنترل میکنند:
- برخی از ژنها که به رشد، تقسیم یا زنده ماندن سلولها کمک میکنند، گاهی اوقات میتوانند به انکوژن (oncogenes) تبدیل شوند.
- ژنهایی که به کنترل تقسیم سلولی، ترمیم DNA آسیب دیده یا مرگ سلولها در زمان مناسب کمک میکنند، ژنهای سرکوبگر تومور (tumor suppressor genes) نامیده میشوند.
سرطانها میتوانند ناشی از تغییرات DNA باشند که انکوژنها را ایجاد میکنند یا ژنهای سرکوب کننده تومور را خاموش میکنند. این تغییرات ژنی گاهی اوقات میتواند از والدین به ارث برسد (که در نوروبلاستوم نادر است) یا ممکن است در طول زندگی فرد اتفاق بیفتد زیرا سلولهای بدن برای ساخت سلولهای جدید تقسیم میشوند.
اغلب، سلولهای نوروبلاستوما دارای تغییرات کروموزومی هستند (مانند داشتن کروموزومهای بسیار زیاد یا کم یا از دست دادن بخشی از کروموزوم) که احتمالاً بر ژنهای خاصی تأثیر میگذارد. دانشمندان هنوز در حال تلاش برای تعیین اینکه کدام ژنها تحت تأثیر این تغییرات کروموزوم قرار میگیرند و همچنین چگونگی تأثیر این تغییرات بر رشد سلولهای نوروبلاستوما، هستند.
تغییرات ژنی اکتسابی (Acquired) در مقابل ارثی (inherited) در نوروبلاستوما
اکثر نوروبلاستومها نتیجه تغییرات ژنی در نوروبلاستها هستند که در طول رشد کودک و گاهی حتی قبل از تولد اتفاق میافتند. علت این تغییرات ژنی اکتسابی مشخص نیست. آنها ممکن است فقط رویدادهای تصادفی باشند که گاهی اوقات در داخل سلولها اتفاق میافتند، بدون اینکه علت بیرونی داشته باشند.
این تغییرات ژنی اکتسابی فقط در سلولهای سرطانی کودک یافت میشود، بنابراین به فرزندانشان منتقل نمیشود.
در موارد نادر، نوروبلاستوما در نتیجه تغییرات ژنی به ارث رسیده از والدین رخ میدهد. تغییرات ارثی در ژنهای خاص بیشتر نوروبلاستوماهای ارثی (خانوادگی) را تشکیل میدهند:
- تغییرات در انکوژن ALK بیشتر موارد نوروبلاستوم ارثی را تشکیل میدهد.
- تغییرات در – ژنی که به طور معمول به بالغ شدن سلولهای عصبی کمک میکند – تعداد کمی از نوروبلاستومهای ارثی را تشکیل میدهد.
برخی از تغییرات ژنی نوروبلاستوما که میتواند بر دیدگاه کودک تأثیر بگذارد
به نظر میرسد برخی تغییرات ژنی دیگر بر سرعت رشد نوروبلاستوما تأثیر میگذارد:
- سلولهای نوروبلاستوما گاهی دارای نسخههای اضافی (تقویتی) از انکوژن MYCN هستند که اغلب نشانهای از رشد سریع تومور است و ممکن است درمان آن سختتر باشد.
- اگر ژن NTRK1 (که پروتئین TrkA را میسازد) در سلولهای نوروبلاستوما بیش فعال باشد، نوروبلاستوم ممکن است چشم انداز بهتری داشته باشد.
- سلولهای نوروبلاستوما در کودکان بزرگتر به احتمال زیاد تغییراتی در ژن سرکوبگر تومور ATRX دارند. تومورهایی با این تغییر ژنی تمایل به رشد آهستهتری دارند اما درمان آنها نیز سختتر است. این ممکن است به توضیح اینکه چرا کودکان کوچکتر مبتلا به نوروبلاستوما در دراز مدت نسبت به کودکان بزرگتر در هنگام تشخیص، عملکرد بهتری دارند، بپردازد.
هنوز مشخص نیست که چه چیزی باعث ایجاد بسیاری از تغییرات ژنی و کروموزومی میشود که میتواند منجر به نوروبلاستوم شود. هیچ دلیل شناخته شده ای برای نوروبلاستوما مرتبط با سبک زندگی یا محیطی وجود ندارد، بنابراین مهم است که به خاطر داشته باشید که این کودکان یا والدین آنها هیچ کاری نمیتوانند برای پیشگیری از این سرطانها انجام دهند.
عوامل خطرزا برای نوروبلاستوما
عامل خطرزا هر چیزی است که شانس ابتلا به بیماری مانند سرطان را افزایش دهد. انواع مختلف سرطان عوامل خطرزا متفاوتی دارند.
عوامل خطرزا مرتبط با سبک زندگی مانند وزن بدن، فعالیت بدنی، رژیم غذایی و استفاده از تنباکو و الکل نقش عمده ای در بسیاری از سرطانهای بزرگسالان دارند اما این عوامل معمولاً سالها طول میکشد تا بر خطر سرطان تأثیر بگذارند و تصور نمیشود که نقش زیادی در سرطانهای دوران کودکی از جمله نوروبلاستوما داشته باشند.
هیچ عامل محیطی (مانند قرار گرفتن در معرض مواد شیمیایی یا تشعشع در دوران بارداری مادر یا در اوایل کودکی) که شانس ابتلا به نوروبلاستوم را افزایش دهد، شناخته شده نیست.
سن
نوروبلاستوما در نوزادان و کودکان بسیار خردسال شایع است. در افراد بالای 10 سال بسیار نادر است.
وراثت
به نظر نمیرسد که اکثر نوروبلاستوماها در خانوادهها دیده شوند اما در حدود 1 تا 2 درصد موارد، کودکان مبتلا به نوروبلاستوما سابقه خانوادگی آن را دارند.
در کودکان مبتلا به این شکل خانوادگی نوروبلاستوما (آنهایی که تمایل ارثی به این سرطان دارند)، میانگین سن در هنگام تشخیص کمتر از سن موارد پراکنده (نه ارثی) است. کودکان مبتلا به نوروبلاستوم خانوادگی گاهی اوقات بیش از یکی از این سرطانها را، اغلب در اندامهای مختلف (مثلاً در هر دو غدد فوق کلیوی یا در بیش از یک گانگلیون سمپاتیک) ایجاد میکنند.
مهم است که نوروبلاستومهایی را که در بیش از یک اندام شروع میشوند از نوروبلاستومهایی که در یک اندام شروع میشوند و سپس به سایرین گسترش مییابند (نوروبلاستومهای متاستاتیک)، متمایز کنیم زیرا تومورهایی که در چندین مکان به طور همزمان ایجاد میشوند احتمالاً خانوادگی هستند. این ممکن است به این معنی باشد که اعضای خانواده باید مشاوره و آزمایش ژنتیک را در نظر بگیرند.
داشتن نقایص مادرزادی (ناهنجاریهای مادرزادی یا congenital anomalies)
برخی از مطالعات نشان داده اند که کودکان با نقصهای مادرزادی خاص ممکن است خطر ابتلا به نوروبلاستوما را افزایش دهند. برخی از ارتباط بین نقایص مادرزادی و نوروبلاستوم ممکن است به تغییرات در ژنهایی که در طول رشد جنین رخ میدهند، مرتبط باشد.
ژنها دستورالعملهایی در هر یک از سلولهای بدن ما هستند که به آنها میگویند که چه کاری انجام دهند. رشد جنین که در رحم مادر اتفاق میافتد، توسط ژنهایی کنترل میشود که به سلولها میگویند چگونه رشد و تقسیم شوند. اگر رشد و تکامل سلولی به طور طبیعی در جنین اتفاق نیفتد، میتواند منجر به نقص مادرزادی شود.
تغییرات در ژنها در طول رشد جنین ممکن است خطر ابتلا به برخی از انواع سرطانهای دوران کودکی مانند نوروبلاستوما را افزایش دهد. برای اطلاعات بیشتر در مورد ژنها و علل نوروبلاستوما، به مبحث چه چیزی باعث نوروبلاستوم میشود؟ مراجعه کنید.
آیا نوروبلاستوما قابل پیشگیری است؟
خطر ابتلا به بسیاری از سرطانهای بزرگسالان را میتوان با تغییرات خاصی در سبک زندگی کاهش داد (مانند حفظ وزن سالم یا ترک سیگار) اما در حال حاضر هیچ راه شناخته شده ای برای پیشگیری از بیشتر سرطانها در کودکان وجود ندارد.
تنها عوامل خطرزا شناخته شده برای نوروبلاستوما قابل تغییر نیستند. هیچ علت شناخته شده ای مرتبط با سبک زندگی یا محیطی برای نوروبلاستوم وجود ندارد.
برخی از مطالعات نشان میدهند که مصرف مولتی ویتامینها یا اسید فولیک در دوران بارداری توسط زنان باردار ممکن است خطر ابتلا به نوروبلاستوما را در فرزندانشان کاهش دهد اما تحقیقات بیشتری برای تایید این موضوع مورد نیاز است.
اگر سابقه نوروبلاستوما در خانواده شما وجود دارد، ممکن است بخواهید با یک مشاور ژنتیک در مورد خطرات ابتلای فرزندانتان به این بیماری صحبت کنید. با این حال، مهم است که به یاد داشته باشید که نوروبلاستوم خانوادگی بسیار نادر است.
آیا نوروبلاستوما در مراحل اولیه تشخیص داده میشود؟
برخی از نوروبلاستومها را میتوان در مراحل اولیه، قبل از اینکه شروع به ایجاد علائم یا نشانهها کنند، پیدا کرد.
به عنوان مثال، تعداد کمی نوروبلاستوم قبل از تولد در طی سونوگرافی یافت میشود، آزمایشی که از امواج صوتی برای ایجاد تصویری از اندامهای داخلی جنین استفاده میکند. سونوگرافی اغلب در دوران بارداری برای تخمین سن جنین، پیش بینی تاریخ تولد و بررسی برخی از نقایص رایج مادرزادی انجام میشود.
همچنین گاهی اوقات نوروبلاستوما به طور تصادفی در کودکان خردسال بدون هیچ علامتی در طی آزمایشاتی که برای بررسی سایر بیماریهای دوران کودکی یا در معاینات منظم پزشکی انجام میشود، مشاهده میشود.
اما اغلب، نوروبلاستوما ابتدا به دلیل علائم یا نشانههایی که کودک دارد، شناسایی میشود.
غربالگری نوروبلاستوما
غربالگری آزمایش بررسی وجود بیماری مانند سرطان در افرادی است که هیچ علامتی ندارند. غربالگری میتواند به یافتن برخی از انواع سرطان در مراحل اولیه کمک کند، زمانی که احتمالاً درمان آنها آسان تر است.
مطالعات در چندین کشور غربالگری نوزادان برای نوروبلاستوم را بررسی کرده اند. غربالگری با آزمایش ادرار نوزادان از نظر مواد شیمیایی خاص ساخته شده توسط تومورهای نوروبلاستوما انجام شد. با این حال، این مطالعات غربالگری نوروبلاستوما را مفید تشخیص ندادند.
غربالگری تومورهای زیادی را پیدا کرد که معمولاً شناسایی نمیشدند اما بیشتر این تومورها احتمالاً به خودی خود از بین میرفتند یا به تومورهای خوش خیم (غیر سرطانی) بالغ میشدند، بنابراین اگر پیدا نمیشدند هرگز مشکلی ایجاد نمیکردند. (برای دریافت اطلاعات بیشتر در این مورد به مبحث نوروبلاستوما چیست؟ مراجعه کنید.) غربالگری تعداد سرطانهای یافت شده در مراحل پیشرفته و همچنین تعداد مرگ و میر ناشی از نوروبلاستوم را کاهش نداد.
غربالگری برای نوروبلاستوما میتواند جنبههای منفی نیز داشته باشد. به عنوان مثال، یافتن تومورهایی که هرگز باعث ایجاد مشکلات جدی نمیشوند، ممکن است بیهوده والدین را بترساند و منجر به انجام آزمایشات و جراحیهای غیر ضروری در برخی از کودکان شوند.
به این دلایل، اکثر متخصصان غربالگری برای نوروبلاستوما را در نوزادانی که در معرض خطر ابتلا به این بیماری نیستند، توصیه نمیکنند.
غربالگری ممکن است برای نوزادانی که در معرض خطر بیشتری هستند، مانند آنهایی که سابقه خانوادگی نوروبلاستوما دارند، توصیه شود. همراه با آزمایش ادرار، این ممکن است شامل آزمایش ژنتیکی برای بررسی تغییرات در ژن ALK نیز باشد که اغلب در موارد نوروبلاستوم ارثی (خانوادگی) دیده میشود.
آزمایشات نوروبلاستوما
نوروبلاستوما معمولاً هنگامی که کودک به دلیل نشانهها یا علائمی که دارد نزد پزشک آورده میشود، پیدا میشود. اگر پزشک به نوروبلاستوما (یا نوع دیگری از تومور) مشکوک باشد، آزمایشاتی برای تایید تشخیص مورد نیاز است.
اگر نوروبلاستوما پیدا شود، آزمایشات دیگری برای کسب اطلاعات بیشتر در مورد آن مورد نیاز خواهد بود.
سابقه پزشکی و معاینه فیزیکی
اگر کودک شما نشانهها یا علائمی داشته باشد که ممکن است ناشی از نوروبلاستوما (یا تومور دیگر) باشد، پزشک در مورد علائم و مدت زمان وجود آنها سوال خواهد کرد. همچنین ممکن است پزشک بپرسد که آیا عوامل خطرزا احتمالی مانند سابقه خانوادگی نوروبلاستوما وجود دارد یا خیر.
پزشک کودک شما را از نظر علائم احتمالی نوروبلاستوما یا سایر مشکلات سلامتی معاینه میکند. به عنوان مثال، پزشک ممکن است شکم را برای یافتن هر گونه توده غیر طبیعی یا تورم معاینه کند. پزشک ممکن است تودهها یا برجستگیهای زیر پوست را بررسی و چشمهای کودک شما را از نزدیک معاینه کند.
آنها همچنین ممکن است فشار خون کودک را بررسی کنند زیرا گاهی اوقات نوروبلاستوم میتواند باعث فشار خون بالا شود. نوروبلاستوما گاهی اوقات میتواند نزدیک نخاع رشد کند که میتواند بر حرکت و قدرت در بازوها و پاهای کودک تأثیر بگذارد، بنابراین پزشک ممکن است به این موارد توجه زیادی داشته باشد.
برخی از علائمی که ممکن است در اثر نوروبلاستوم ایجاد شوند، مانند تب و بزرگ شدن غدد لنفاوی، به احتمال زیاد ناشی از عفونت هستند، بنابراین پزشک ممکن است در ابتدا به دنبال علائم دیگر عفونت باشد.
اگر شرح حال و معاینه نشان دهد که کودک ممکن است نوروبلاستوما (یا نوع دیگری از تومور) داشته باشد، آزمایشهای بیشتری انجام خواهد شد. این موارد میتواند شامل موارد زیر باشد:
- آزمایش خون و ادرار
- تستهای تصویر برداری
- بیوپسی (نمونه برداری)
این آزمایشها مهم هستند زیرا بسیاری از علائم و نشانههای نوروبلاستوما میتوانند ناشی از بیماریهای دیگر مانند عفونتها یا حتی انواع دیگر سرطان باشند.
آزمایشات کاتکول آمین خون و ادرار
سلولهای بدن انواع مختلفی از هورمونها را میسازند. به عنوان مثال، سلولهای عصبی سمپاتیک به طور معمول هورمونهایی به نام کاتکول آمینها (catecholamines) مانند اپی نفرین (آدرنالین) و نوراپی نفرین را آزاد میکنند که وارد خون شده و در نهایت به قطعات کوچک تری به نام متابولیتها تجزیه میشوند.

متابولیتها معمولاً از طریق ادرار از بدن خارج میشوند. هنگامی که اپی نفرین و نوراپی نفرین توسط بدن تجزیه میشوند، دو متابولیت رایج ساخته شده عبارتند از:
- هومووانیلیک اسید (Homovanillic acid یا HVA)
- وانیللماندلیک اسید (Vanillylmandelic acid یا VMA)
سلولهای نوروبلاستوما نیز اغلب این کاتکول آمینها را میسازند، بنابراین همین متابولیتها را میتوان در خون و ادرار تشخیص داد. اگر سلولهای نوروبلاستوما در حال ساخت کاتکول آمین باشند، میزان HVA و VMA در ادرار یا خون بیشتر از حد انتظار خواهد بود.
اگر کودکی نوروبلاستوما داشته باشد، سطوح HVA و VMA را نیز میتوان در طول دوره درمان دنبال کرد تا از عملکرد آن مطلع شد.
سایر آزمایشات خون و ادرار: اگر نوروبلاستوما مشکوک باشد یا پیدا شده باشد، پزشک کودک شما احتمالاً آزمایش خون را برای بررسی تعداد سلولهای خونی، عملکرد کبد و کلیه و تعادل املاح (الکترولیتها) در بدن تجویز میکند. آزمایش ادرار (urine test) نیز ممکن است برای کمک به بررسی عملکرد کلیه انجام شود.
تستهای تصویر برداری
تستهای تصویر برداری از اشعه ایکس، میدانهای مغناطیسی، امواج صوتی یا مواد رادیواکتیو برای ایجاد تصاویری از داخل بدن استفاده میکنند. تستهای تصویر برداری را میتوان به دلایل مختلفی انجام داد، از جمله:
- برای کمک به یافتن اینکه آیا یک ناحیه مشکوک ممکن است سرطان باشد یا خیر.
- برای فهم میزان گسترش سرطان
- برای کمک به تعیین اینکه آیا روند درمان موثر است یا خیر.
اکثر کودکانی که نوروبلاستوما دارند یا ممکن است داشته باشند، یک یا چند مورد از این آزمایشها را انجام میدهند اما ممکن است به همه آنها نیاز نداشته باشند.
کودکان مبتلا به نوروبلاستوما اغلب بسیار جوان هستند، بنابراین انجام برخی از این آزمایشات ممکن است سخت باشد زیرا ممکن است کودک نیاز به ثابت نگه داشتن داشته باشد. بسته به سن کودک و آزمایش تصویر برداری در حال انجام، ممکن است کودک داروهایی را برای خواب آلوده شدن (یا حتی فرو رفتن در خواب عمیق) دریافت کنند تا به او کمک کند بیحرکت بماند.
سونوگرافی (Ultrasound)
اولتراسوند از امواج صوتی و پژواک آنها برای مشاهده داخل بدن استفاده میکند. این ممکن است یکی از اولین آزمایشهایی باشد که در صورت مشکوک شدن به تومور در کودکان بسیار کوچک انجام میشود زیرا نسبتاً سریع و آسان است، از اشعه استفاده نمیکند و اغلب میتواند دید خوبی از داخل بدن به پزشک بدهد، به خصوص در شکم (Ultrasound). اگر کودک قبلاً ام آر آی یا سی تی اسکن انجام داده باشد معمولاً سونوگرافی لازم نیست.
سونوگرافی اغلب برای جستجوی تومور در شکم استفاده میشود. (برای نگاه کردن به قفسه سینه استفاده نمیشود زیرا دندهها امواج صوتی را مسدود میکنند.)
تصویر برداری رزونانس مغناطیسی (MRI یا Magnetic resonance imaging)
اسکن MRI تصاویر دقیقی از بافتهای نرم بدن ارائه میدهد. این اسکنها ممکن است کمی بهتر از سی تی اسکن برای مشاهده وسعت تومور نوروبلاستوما، به ویژه در اطراف ستون فقرات باشد.
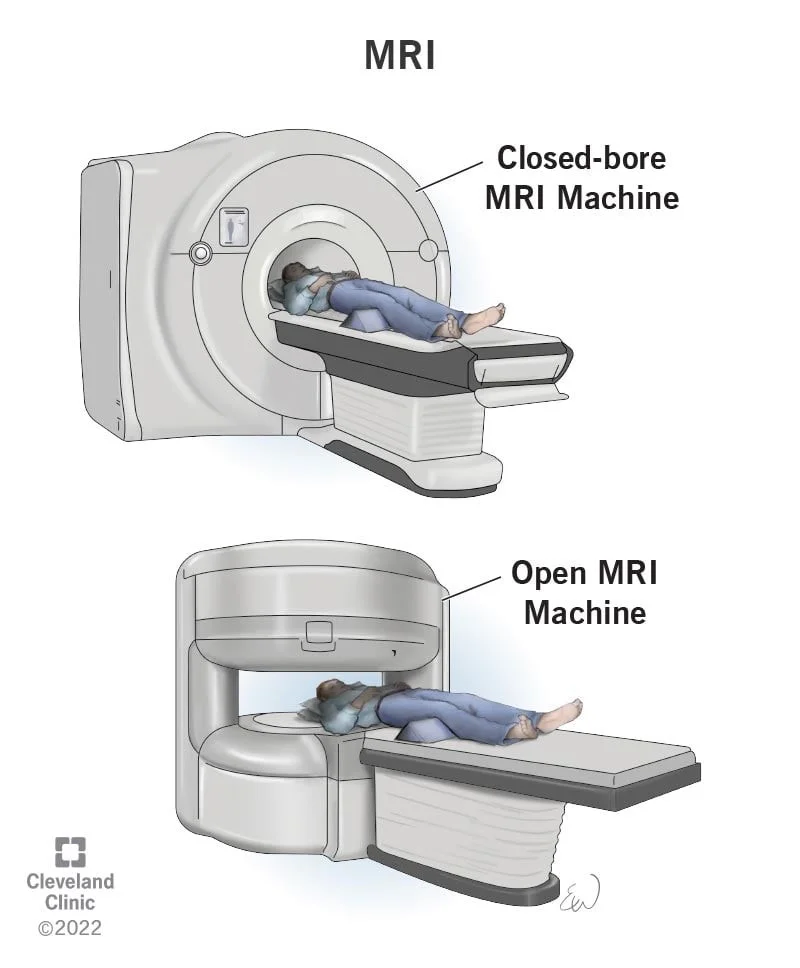
اسکنهای MRI از امواج رادیویی و آهنرباهای قوی برای ایجاد تصاویر به جای اشعه ایکس استفاده میکنند، بنابراین هیچ تشعشعی وجود ندارد. ماده حاجب به نام گادولینیوم (gadolinium) ممکن است قبل از اسکن به داخل ورید تزریق شود تا جزئیات را بهتر ببینند.
اسکن توموگرافی کامپیوتری (CT یا Computed tomography یا CAT)
سی تی اسکن بسیاری از تصاویر اشعه ایکس را برای ایجاد تصاویر مقطعی دقیق از داخل بدن ترکیب میکند. این آزمایشات اغلب برای بررسی نوروبلاستوما در شکم، لگن و قفسه سینه استفاده میشود.
قبل از انجام آزمایش، ممکن است از کودک شما خواسته شود که محلول کنتراست (contrast solution) را بنوشد و یا تزریق IV (داخل وریدی) رنگ کنتراست انجام دهد. این به ترسیم بهتر ساختارهای بدن کمک میکند.
بیوپسی سوزنی با هدایت CT (CT-guided needle biopsy): از سی تی اسکن میتوان برای کمک به هدایت سوزن بیوپسی به داخل تومور نیز استفاده کرد. اما در صورت مشکوک شدن به نوروبلاستوم، نمونه برداری سوزنی (که در زیر توضیح داده شده است) اغلب انجام نمیشود زیرا مقدار نمونه جمع آوری شده ممکن است برای تمام آزمایشهای تومور مورد نیاز کافی نباشد.
اسکن MIBG
این آزمایش اغلب بخش مهمی برای یافتن میزان گسترش نوروبلاستوما در کودک است. اغلب پس از انجام سی تی اسکن یا ام آر آی انجام میشود.
برای این آزمایش، نوعی از ماده شیمیایی متا یدوبنزیل گوانیدین (meta-iodobenzylguanidine یا MIBG) که حاوی مقدار کمی ید رادیواکتیو است به خون تزریق میشود. MIBG شبیه نوراپی نفرین است، هورمونی که توسط سلولهای عصبی سمپاتیک ساخته میشود و در اکثر بیماران به سلولهای نوروبلاستوما در هر نقطه از بدن متصل میشود. بین 1 تا 3 روز بعد، بدن با یک دوربین ویژه اسکن میشود تا به دنبال مناطقی باشد که رادیواکتیو را دریافت کرده است. این به پزشکان کمک میکند تا بدانند نوروبلاستوم کجاست و آیا به استخوانها و یا سایر قسمتهای بدن گسترش یافته است یا خیر.
اسکن MIBG را میتوان پس از درمان تکرار کرد تا ببینند آیا تومورها به خوبی پاسخ میدهند یا خیر. همچنین خوب است بدانید که آیا تومور MIBG را میگیرد یا خیر، زیرا در برخی موارد، این مولکول رادیواکتیو را میتوان در دوزهای بالاتر برای درمان نوروبلاستوم استفاده کرد.
غده تیروئید همچنین میتواند MIBG را جذب کند، بنابراین گاهی اوقات یک داروی حاوی ید قبل، حین و بعد از آزمایش برای محافظت از تیروئید تجویز میشود.
اسکن استخوان (Bone scan)

اسکن استخوان میتواند نشان دهد که آیا سرطان به استخوانها گسترش یافته است یا خیر و میتواند تصویری از کل اسکلت را به یکباره ارائه دهد. نوروبلاستوما اغلب باعث آسیب استخوان میشود که در اسکن استخوان قابل مشاهده است.
این آزمایش اغلب برای نوروبلاستوما مورد نیاز نیست زیرا اسکن MIBG معمولاً میتواند گسترش سرطان به استخوان را تشخیص دهد اما اگر اسکن MIBG سرطان را در استخوان پیدا نکرد و پزشک همچنان مشکوک است که ممکن است در آن جا گسترش یافته باشد، اسکن استخوان ممکن است مفید باشد.
برای این آزمایش، مقدار کمی از مواد رادیواکتیو سطح پایین (تکنسیوم-99) به داخل ورید تزریق میشود. (میزان رادیواکتیویته مورد استفاده بسیار کم است و در عرض یک روز یا بیشتر از بدن خارج میشود.) این ماده در طی چند ساعت در نواحی استخوان آسیب دیده در سراسر اسکلت مینشیند. سپس کودک شما حدود 30 دقیقه روی میز دراز میکشد در حالی که یک دوربین مخصوص رادیواکتیویته را تشخیص داده و تصویری از اسکلت ایجاد میکند. ممکن است به کودکان کوچکتر دارو داده شود تا در طول آزمایش آرام بمانند یا حتی بخوابند.
نواحی تغییرات استخوانی فعال، رادیواکتیویته را جذب کرده و به صورت “نقاط داغ یا hot spots” روی اسکلت ظاهر میشوند. این مناطق ممکن است نشان دهنده سرطان باشند اما سایر بیماریهای استخوانی نیز میتوانند همین الگو را ایجاد کنند. برای کمک به تشخیص این موارد، آزمایشهای تصویر برداری دیگری مانند عکس برداری با اشعه ایکس ساده یا اسکن MRI یا حتی بیوپسی استخوان ممکن است مورد نیاز باشد.
اسکن توموگرافی گسیل پوزیترون (PET یا Positron emission tomography)
برای اسکن PET، یک ماده رادیواکتیو (معمولا نوعی قند به نام FDG) به خون تزریق میشود. میزان رادیواکتیویته مورد استفاده بسیار کم است و در عرض یک روز یا بیشتر از بدن خارج میشود.
از آن جایی که سلولهای سرطانی به سرعت در حال رشد هستند، مقادیر زیادی از قند رادیواکتیو را جذب میکنند. پس از حدود یک ساعت، کودک شما روی یک میز در اسکنر PET منتقل میشود. آنها حدود 30 دقیقه روی میز دراز میکشند در حالی که یک دوربین ویژه تصویری را از مناطق پرتوزا در بدن ایجاد میکند. ممکن است به کودکان کوچکتر دارو داده شود تا در طول آزمایش آرام بمانند یا حتی بخوابند.
اگر اسکن MIBG انجام شده باشد معمولاً اسکن PET لازم نیست اما اسکن PET ممکن است برای برخی از نوروبلاستومها مفید باشد، به خصوص اگر سلولهای نوروبلاستوم MIBG را جذب نکنند.
اشعه ایکس (X-rays)
از اشعه ایکس میتوان برای مشاهده استخوانها استفاده کرد، اگرچه آنها در نشان دادن سایر ساختارهای بدن به این خوبی نیستند.
در صورتی که کودک علائمی داشته باشد و مشخص نباشد که چه چیزی ممکن است باعث آن شود، ممکن است پزشک به عنوان یک آزمایش اولیه، عکس برداری از قسمتی از بدن را با اشعه ایکس تجویز کند اما این تصاویر ممکن است برای تشخیص تومورها کافی نباشد.
در کودکان مبتلا به نوروبلاستوم، MIBG، PET یا اسکن استخوان معمولاً بهتر از اشعه ایکس برای بررسی استخوانهای بقیه بدن و بررسی اینکه آیا نوروبلاستوم به استخوانها گسترش یافته است یا خیر، عمل میکنند اما آزمایش اشعه ایکس نیز ممکن است هنوز هم در برخی شرایط مفید باشد.
بیوپسیها (Biopsies)
در طول بیوپسی، پزشک یک یا چند قطعه (نمونه) را برای آزمایش از تومور خارج میکند.
معاینات و آزمایشات تصویر برداری ممکن است به شدت نشان دهد که یک کودک مبتلا به نوروبلاستوم است اما معمولاً برای اطمینان از بیوپسی لازم است. (برخی از نوزادان بسیار جوان با تومورهای کوچک آدرنال که در آزمایش تصویر برداری مشاهده میشوند، ممکن است نیازی به بیوپسی نداشته باشند. در عوض، تومور ممکن است با آزمایشهای تصویر برداری بیشتر به دقت زیر نظر گرفته شود زیرا این تومورها اغلب بالغ میشوند یا خود به خود از بین میروند.)
در بزرگسالان، بیوپسی گاهی با استفاده از بی حس کننده موضعی (داروی بی حسی) انجام میشود اما در کودکان اغلب در حالی که کودک تحت بیهوشی عمومی است (در خواب) انجام میشود.
2 نوع اصلی بیوپسی وجود دارد:
- بیوپسی برشی یا Incisional biopsy (باز یا جراحی): این رایج ترین نوع بیوپسی برای نوروبلاستوم است. این کار با برداشتن قطعه ای از تومور از طریق یک برش (بریدگی) در پوست انجام میشود. برای تومورهای موجود در اعماق بدن، این ممکن است به روش لاپاراسکوپی (laparoscopically) با استفاده از ابزارهای جراحی بلند و نازک که از طریق بریدگیهای کوچک در پوست وارد میشوند، انجام شود.
- بیوپسی سوزنی یا Needle biopsy (بسته): برای این نوع بیوپسی، یک سوزن نازک و توخالی از طریق پوست وارد تومور میشود تا نمونه کوچکی از آن خارج شود. اگر تومور در عمق بدن باشد، میتوان از سی تی اسکن یا سونوگرافی برای هدایت سوزن به داخل تومور استفاده کرد. بیوپسی سوزنی معمولاً زمانی که کودک ممکن است نوروبلاستوما داشته باشد، مفید نیست زیرا مقدار تومور در نمونه اغلب برای انجام تمام آزمایشهای ویژه مورد نیاز کافی نیست.
پس از برداشتن نمونههای بیوپسی، آنها به آزمایشگاه فرستاده میشوند و در زیر میکروسکوپ توسط پاتولوژیست (پزشک با آموزش ویژه در شناسایی سلولهای سرطانی) مشاهده میشوند. تستهای آزمایشگاهی ویژه اغلب بر روی نمونهها نیز انجام میشود تا نشان دهد که آیا تومور نوروبلاستوما است یا خیر.
اگر این یک نوروبلاستوما باشد، تستهای آزمایشگاهی همچنین میتوانند به تعیین سرعت رشد یا گسترش تومور و همچنین اینکه کدام درمانها ممکن است بهترین کار را انجام دهند، کمک کنند. برخی از این تستها در مبحث مراحل نوروبلاستوما و نشانگرهای پیش آگهی توضیح داده شده اند.
آسپیراسیون مغز استخوان (Bone marrow aspiration) و بیوپسی
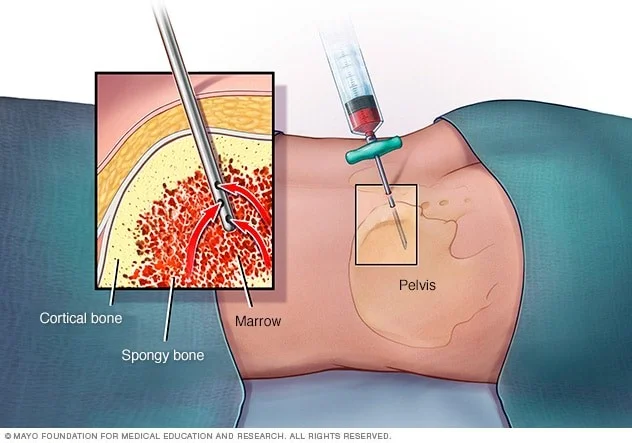
نوروبلاستوما اغلب به مغز استخوان (بخشهای داخلی نرم برخی از استخوانها) گسترش مییابد. اگر سطح کاتکول آمینها در خون یا ادرار افزایش یابد، یافتن سلولهای سرطانی در نمونه مغز استخوان برای تشخیص نوروبلاستوم (بدون بیوپسی از تومور اصلی) کافی است. اگر نوروبلاستوما قبلاً با بیوپسی انجام شده در جای دیگری از بدن تشخیص داده شده باشد، آزمایشات مغز استخوان برای کمک به تعیین وسعت بیماری انجام میشود.
آسپیراسیون مغز استخوان و بیوپسی معمولاً همزمان انجام میشود. در اغلب موارد نمونهها از پشت هر دو استخوان لگن (hip) گرفته میشود.
حتی زمانی که ناحیه با بی حسی موضعی بی حس میشود، این آزمایشها میتوانند دردناک باشند، بنابراین در بیشتر موارد به کودک داروهای دیگری برای کاهش درد یا حتی خوابیدن در طول عمل داده میشود.
در آسپیراسیون مغز استخوان، یک سوزن نازک و توخالی به استخوان وارد شده و از یک سرنگ برای مکیدن مقدار کمی از مغز استخوان مایع استفاده میشود.
برای بیوپسی مغز استخوان، یک تکه کوچک از استخوان و مغز استخوان با یک سوزن کمی بزرگتر که به پایین به داخل استخوان فشار داده میشود، برداشته میشود. پس از انجام بیوپسی، فشار به محل برای کمک به توقف هرگونه خونریزی وارد میشود.
نمونههایی از مغز استخوان به آزمایشگاه فرستاده شده و در آن جا بررسی و برای وجود سلولهای سرطانی آزمایش میشوند.
مراحل نوروبلاستوما و نشانگرهای پیش آگهی
اگر فردی مبتلا به نوروبلاستوما تشخیص داده شود، پزشکان سعی میکنند بفهمند که آیا گسترش یافته است یا خیر و اگر چنین است، این گسترش تا کجاست. این فرآیند مرحله بندی (staging) نامیده میشود. مرحله نوروبلاستوما میزان سرطان در بدن را توصیف میکند.
(برای نوروبلاستوما، چندین عامل دیگر همراه با مرحله کودک بررسی میشود تا تصمیم بگیرند که کودک در کدام گروه خطر قرار میگیرد. گروههای خطر یک تصویر کلی از چگونگی پاسخ احتمالی نوروبلاستوما به روند درمان ارائه میدهند و به پزشکان کمک میکنند تا درمانهایی را انتخاب کنند که ممکن است بهترین کار را انجام دهند. پزشکان همچنین هنگام صحبت در مورد آمار بقا از گروههای خطر نوروبلاستوما استفاده میکنند. برای اطلاعات بیشتر، به مبحث گروههای خطر نوروبلاستوما مراجعه کنید.)
دو سیستم برای مرحله بندی نوروبلاستوم استفاده میشود. تفاوت اصلی بین آنها این است که آیا میتوان از سیستم مرحله بندی برای کمک به تعیین گروه خطر کودک قبل از شروع درمان استفاده کرد.
- سیستم بین المللی مرحله بندی گروه خطر نوروبلاستوما (International Neuroblastoma Risk Group Staging System یا INRGSS) از نتایج آزمایشهای تصویر برداری (مانند CT یا MRI و اسکن MIBG) برای کمک به تصمیم گیری در مورد مرحله استفاده میکند. مرحله INRGSS را میتوان قبل از شروع درمان تعیین کرد.
- سیستم بین المللی مرحله بندی نوروبلاستوما (INSS یا International Neuroblastoma Staging System) بر اساس نتایج حاصل از جراحی برداشتن تومور کودک به جای آزمایشهای تصویر برداری است.
این سیستمهای مرحله بندی میتوانند هر دو برای کمک به اطمینان یافتن از اینکه کودکان مبتلا به نوروبلاستوما بهترین درمانها را دریافت میکنند، استفاده شوند. اگر فرزند شما نوروبلاستوما داشته باشد و جراحی نکرده باشد، به احتمال زیاد بر اساس INRGSS در مورد مرحله کودک خود خواهید شنید. اگر فرزند شما جراحی شده باشد، ممکن است با استفاده از هر یک از این سیستمها، پزشکان در مورد مرحله کودک خود صحبت کنند.
مراحل نوروبلاستوم پیچیده است و ممکن است گیج کننده باشد. اگر در مورد معنای آنها برای کودک خود مطمئن نیستید، از پزشک کودک خود بخواهید که آنها را به گونه ای که قابل درک باشد برای شما توضیح دهد.
سیستم مرحله بندی بینالمللی گروه خطر نوروبلاستوما (INRGSS)
INRGSS برای کمک به تعیین مرحله کودک و گروه خطر قبل از شروع دوره درمان ایجاد شده است. همچنین به محققان در سراسر جهان کمک کرده است تا نتایج مطالعات را با هم مقایسه کنند تا بفهمند کدام درمان بهترین روش است. INRGSS از تستهای تصویر برداری (معمولاً اسکن CT یا MRI و اسکن MIBG) و همچنین از معاینه و بیوپسی برای کمک به تعریف مرحله استفاده میکند. سپس میتوان از این مرحله برای کمک به پیش بینی میزان قابل برداشتن تومور استفاده کرد – یعنی اینکه چه مقدار از آن را میتوان با جراحی برداشت.
INRGSS از عوامل خطرزا تعریف شده با تصویر (image-defined risk factors یا IDRFs) استفاده میکند که عواملی هستند که در آزمایشهای تصویر برداری مشاهده میشوند و ممکن است به این معنی باشند که برداشتن تومور دشوارتر خواهد بود. این شامل مواردی مانند رشد تومور به یک اندام حیاتی نزدیک یا رشد در اطراف عروق خونی مهم است.
INRGSS نوروبلاستومها را به 4 مرحله تقسیم میکند:
L1: تومور از جایی که شروع شده است گسترش نیافته است و به ساختارهای حیاتی که در لیست IDRFها تعریف شده است، رشد نکرده است. این بیماری به یک ناحیه از بدن مانند گردن، سینه یا شکم محدود میشود.
L2: تومور دور از جایی که شروع شده گسترش نیافته است (مثلاً ممکن است از سمت چپ شکم به سمت چپ قفسه سینه رشد کرده باشد) اما حداقل یک IDRF دارد.
M: تومور به نقاط دور تر بدن (به جز تومورهایی که در مرحله MS هستند) گسترش یافته است (متاستاز داده است).
MS: بیماری متاستاتیک در کودکان کمتر از 18 ماه که سرطان فقط به پوست، کبد و یا مغز استخوان گسترش مییابد.
سیستم بین المللی مرحله بندی نوروبلاستوما (INSS)
INSS نتایج جراحی برای برداشتن تومور را در نظر میگیرد. این نمیتواند به پزشکان کمک کند تا قبل از شروع هر درمانی مرحله ای را تعیین کنند، بنابراین برای کودکانی که نیازی به جراحی ندارند یا نمیتوانند عمل کنند، به خوبی کار نمیکند. به صورت ساده، مراحل عبارتند از:
مرحله 1: سرطان هنوز در منطقه ای است که شروع شده است. در یک طرف بدن (راست یا چپ) قرار دارد. تمام تومورهای قابل مشاهده به طور کامل توسط جراحی برداشته شده است (اگرچه نگاه کردن به لبههای تومور در زیر میکروسکوپ پس از جراحی ممکن است برخی از سلولهای سرطانی را نشان دهد). گرههای لنفاوی نزدیک تومور عاری از سرطان هستند (اگرچه گرههای محصور در تومور ممکن است حاوی سلولهای نوروبلاستوما باشند).
مرحله 2A: سرطان هنوز در ناحیه شروع و در یک طرف بدن است اما تمام تومور قابل مشاهده با جراحی قابل برداشتن نیست. گرههای لنفاوی نزدیک تومور عاری از سرطان هستند (اگرچه گرههای محصور در تومور ممکن است حاوی سلولهای نوروبلاستوما باشند).
مرحله 2B: سرطان در یک طرف بدن است و ممکن است با جراحی کاملاً برداشته شده باشد یا نشده باشد. غدد لنفاوی مجاور خارج از تومور حاوی سلولهای نوروبلاستوما هستند اما سرطان به غدد لنفاوی در طرف دیگر بدن یا جاهای دیگر گسترش نیافته است.
مرحله 3: سرطان به نقاط دور تر بدن گسترش نیافته است اما یکی از موارد زیر صادق است:
- سرطان را نمیتوان به طور کامل با جراحی برداشت و از خط وسط (که به عنوان ستون فقرات تعریف میشود) به طرف دیگر بدن عبور کرده است. ممکن است به غدد لنفاوی مجاور سرایت کرده باشد یا نه.
- سرطان هنوز در ناحیه ای است که شروع شده و در یک طرف بدن قرار دارد. این بیماری به غدد لنفاوی نسبتاً نزدیک اما در طرف دیگر بدن گسترش یافته است.
- سرطان در وسط بدن قرار دارد و به سمت هر دو طرف در حال رشد است (یا مستقیماً یا با انتشار به غدد لنفاوی مجاور).
مرحله 4: سرطان به قسمتهای دور بدن مانند غدد لنفاوی دور، استخوانها، کبد، پوست، مغز استخوان یا سایر اندامها گسترش یافته است (اما کودک معیارهای مرحله S4 را ندارد).
مرحله S4 (که نوروبلاستوما “ویژه یا special” نیز نامیده میشود): کودک کمتر از 1 سال دارد.
سرطان در یک طرف بدن است. ممکن است به غدد لنفاوی در همان سمت بدن گسترش یافته باشد اما به گرههای طرف دیگر گسترش نیافته است. نوروبلاستوما به کبد، پوست و یا مغز استخوان گسترش یافته است. با این حال، بیش از 10 درصد از سلولهای مغز استخوان را سلولهای سرطانی تشکیل نمیدهند و آزمایشهای تصویر برداری مانند اسکن MIBG سرطان را در مغز استخوان نشان نمیدهند.
عود کننده (Recurrent): در حالی که بخشی رسمی از سیستم مرحله بندی نیست، این اصطلاح برای توصیف سرطانی استفاده میشود که پس از درمان عود کرده است (برگشته است). سرطان ممکن است در ناحیه ای که برای اولین بار شروع شده یا در قسمت دیگری از بدن عود کند.
نشانگرهای پیش آگهی
نشانگرهای پیش آگهی ویژگیهایی هستند که به پیش بینی اینکه آیا پیش آگهی کودک (چشم انداز درمان) بهتر یا بدتر از آن چه در مرحله به تنهایی پیش بینی میشود، کمک میکند. بسیاری از این نشانگرهای پیش آگهی همراه با مرحله کودک برای تعیین گروه خطر استفاده میشوند:
- سن: کودکان کوچکتر (زیر 18-12 ماه) نسبت به کودکان بزرگتر احتمال بیشتری دارد که نتیجه بهتری داشته باشند.
- بافت شناسی تومور: بافت شناسی تومور نحوه ظاهر سلولهای نوروبلاستوما در زیر میکروسکوپ است. تومورهایی که حاوی سلولها و بافتهایی با ظاهر طبیعی بیشتری هستند، پیش آگهی بهتری دارند و گفته میشود که بافت شناسی مطلوبی دارند. تومورهایی که سلولها و بافتهای آنها در زیر میکروسکوپ غیر طبیعیتر به نظر میرسند، پیش آگهی ضعیفتری دارند و گفته میشود که بافت شناسی نامطلوبی دارند.
- DNA پلوئیدی: مقدار DNA در هر سلول که به پلوئیدی یا شاخص DNA معروف است، با استفاده از تستهای آزمایشگاهی خاص قابل اندازه گیری است. سلولهای نوروبلاستوما با تقریباً همان مقدار DNA سلولهای طبیعی (شاخص DNA 1) به عنوان دیپلوئید (diploid) طبقه بندی میشوند. به سلولهایی که مقدار DNA آنها افزایش یافته است (شاخص DNA بالاتر از 1) هیپردیپلوئید (hyperdiploid) میگویند. سلولهای نوروبلاستوما با DNA بیشتر با پیش آگهی بهتری همراه است، به ویژه برای کودکان زیر 2 سال. پلوئیدی DNA برای درک پیش آگهی در کودکان بزرگتر مفید نیست.
- تکثیر ژن MYCN : MYCN ژنی است که به طور معمول به تنظیم رشد سلول کمک میکند. تغییرات در ژن MYCN میتواند آن را به یک انکوژن تبدیل کند که میتواند سلولها را مانند سلولهای سرطانی خیلی سریع رشد و تقسیم کند. نوروبلاستوما با تعداد زیادی کپی (تکثیر) از انکوژن MYCN به سرعت رشد میکند و درمان آن دشوارتر است.
- تغییرات کروموزومی: سلولهای توموری که بخشهای خاصی از کروموزومهای 1 یا 11 را ندارند (که به عنوان حذفهای 1p یا حذفهای 11q شناخته میشود) ممکن است پیش آگهی کمتری را پیش بینی کنند. داشتن یک قسمت اضافی از کروموزوم 17 (افزایش 17q) نیز با پیش آگهی بدتری مرتبط است. درک اهمیت حذف یا افزایش کروموزوم یک حوزه فعال در تحقیقات نوروبلاستوما است.
- گیرندههای نوروتروفین یا Neurotrophin receptors (فاکتور رشد عصبی): اینها موادی هستند که روی سطح سلولهای عصبی طبیعی و روی برخی از سلولهای نوروبلاستوما قرار دارند. آنها معمولاً به سلولها اجازه میدهند نوروتروفینها را که مواد شیمیایی هورمون مانندی هستند که به بلوغ سلولهای عصبی کمک میکنند، تشخیص دهند. نوروبلاستوماهایی که تعداد بیشتری گیرنده نوروتروفین خاص، به ویژه گیرنده فاکتور رشد عصبی TrkA دارند، ممکن است پیش آگهی بهتری داشته باشند.
سطوح سرمی (خون) برخی از مواد نیز میتواند برای کمک به پیش بینی پیش آگهی استفاده شود.
- سلولهای نوروبلاستوما فریتین (ferritin)، یک ماده شیمیایی که بخش مهمی از متابولیسم طبیعی آهن بدن است را در خون آزاد میکنند. بیمارانی که سطح فریتین بالایی دارند پیش آگهی بدتری دارند.
- افزایش سطح لاکتات دهیدروژناز (lactate dehydrogenase یا LDH) در خون نیز با چشم انداز بدتری در کودکان مبتلا به نوروبلاستوما مرتبط است.
گروههای خطر نوروبلاستوما
از گروههای خطر برای کمک به پیش بینی اینکه کودک مبتلا به نوروبلاستوما چقدر احتمال دارد که درمان شود (و بنابراین درمان فشرده چقدر ممکن است نیاز باشد)، استفاده میشود. به عنوان مثال، یک کودک در یک گروه کم خطر اغلب میتواند با درمان محدود، مانند جراحی به تنهایی، درمان شود. کودکان در گروههای پرخطر اغلب به درمان فشردهتری نیاز دارند تا بهترین شانس درمان را داشته باشند.
گروههای خطر بر اساس مرحله (وسعت) سرطان و همچنین سایر عواملی مشخص میشوند که میتوانند بر پیش آگهی (دیدگاه) کودک تأثیر بگذارند، مانند سن.
گروههای خطر که در اینجا گنجانده شده اند، معمولاً گروههای ریسک استاندارد پذیرفته شده در ایالات متحده هستند. سایر گروههای خطر بین المللی مورد استفاده در آزمایشات بالینی در حال آزمایش هستند.
گروههای خطر گروه انکولوژی کودکان (Children’s Oncology Group یا COG)
سیستم گروه خطر گروه انکولوژی کودکان (COG، گروه عمده پزشکانی که سرطان دوران کودکی را در ایالات متحده درمان میکنند) ابتدا بر اساس سیستم مرحله بندی بینالمللی نوروبلاستوما (INSS) بود اما اکنون در حال انتقال به استفاده از مرحله بندی بینالمللی گروه خطر نوروبلاستوما است. سیستم (INRGSS)، همراه با فاکتورهای اصلی پیش آگهی در مبحث مراحل نوروبلاستوما و نشانگرهای پیش آگهی توضیح داده شده اند. اینها برای قرار دادن کودکان در 3 گروه خطر مختلف ترکیب میشوند:
- ریسک کم
- ریسک متوسط
- ریسک بالا
این گروههای خطر بر اساس آن چه در مورد نوروبلاستوما شناخته شده است و نحوه درمان آنها هستند. همان طور که تحقیقات جدید اطلاعات بیشتری را ارائه میدهد، گروههای خطر ممکن است در طول زمان تغییر کنند.
برای مثال، در توصیههای درمانی اخیر، محدودیت سنی برای برخی از این دستهها از 12 ماهگی به 18 ماهگی بازنگری شده است.
طبقه بندی بین المللی گروه خطر نوروبلاستوما (INRG)
طبقه بندی بین المللی گروه خطر نوروبلاستوما (INRG) سیستم جدیدتری است که اکنون برای کمک به محققان در کشورهای مختلف در مقایسه نتایج و همکاری با یکدیگر برای یافتن بهترین درمانها استفاده میشود. این سیستم مبتنی بر سیستم مرحله بندی INRGSS است که شامل عوامل خطرزا تعریف شده با تصویر (IDRFs) و همچنین بسیاری از عوامل پیش آگهی فهرست شده در مراحل نوروبلاستوما و نشانگرهای پیش آگهی است، مانند:
- سن کودک
- بافت شناسی تومور (چگونه تومور در زیر میکروسکوپ به نظر میرسد)
- وجود یا عدم وجود تکثیر ژن MYCN در سلولهای تومور
- تغییرات خاصی در کروموزوم 11 (معروف به انحراف 11q) در سلولهای تومور
- پلوئیدی DNA (تعداد کل کروموزومها در سلولهای تومور)
طبقه بندی INRG از این عوامل برای قرار دادن کودکان در 16 گروه مختلف قبل از درمان (حروف A تا R) استفاده میکند. هر گروه قبل از درمان، به 1 مورد از 4 گروه خطر کلی تقسیم میشود:
- خطر بسیار کم
- ریسک کم
- ریسک متوسط
- ریسک بالا
این سیستم به احتمال زیاد علاوه بر سیستم طبقه بندی ریسک COG در ایالات متحده مورد استفاده قرار خواهد گرفت.
گروههای خطر برای نوروبلاستوم پیچیده هستند و میتوانند گیج کننده باشند. اگر در مورد گروه خطر کودک خود و معنای آن مطمئن نیستید، از پزشک کودکتان بخواهید آن را به گونه ای که قابل درک باشد برای شما توضیح دهد.
نرخ بقای نوروبلاستوما بر اساس گروه خطر
نرخ بقا راهی برای دریافت ایده ای از چشم انداز کودکان مبتلا به نوع خاصی از سرطان است. آنها نمیتوانند به طور قطعی به شما بگویند که آیا درمان موفقیت آمیز خواهد بود یا خیر اما ممکن است به شما کمک کنند تا درک بهتری از احتمال این موضوع به شما بدهند.
میزان بقای 5 ساله چیست؟
نرخ بقای 5 ساله به درصد کودکانی اطلاق میشود که حداقل 5 سال پس از تشخیص سرطان زندگی میکنند. البته، بسیاری از کودکان بسیار بیشتر از 5 سال عمر میکنند (و بسیاری از آنها درمان میشوند).
برای به دست آوردن نرخ بقای 5 ساله، پزشکان باید کودکانی را که حداقل 5 سال پیش تحت درمان قرار گرفته اند، بررسی کنند. بهبود در درمان از آن زمان ممکن است منجر به چشم انداز بهتر برای کودکانی شود که اکنون مبتلا به نوروبلاستوما هستند.
میزان بقا بر اساس نتایج قبلی تعداد زیادی از افرادی است که به این بیماری مبتلا بودند اما آنها نمیتوانند پیش بینی کنند که در مورد کودک خاصی چه اتفاقی خواهد افتاد.
نرخ بقای زیر بر اساس گروه خطر ابتلا به سرطان کودک است. گروه خطر، به نوبه خود، بر اساس مرحله (وسعت) سرطان و همچنین سایر عوامل پیش آگهی (مانند سن کودک) است اما عوامل دیگری نیز میتوانند بر دیدگاه کودک تأثیر بگذارند، مانند محل تومور و اینکه سرطان چگونه به درمان پاسخ میدهد. پزشک فرزندتان میتواند به شما بگوید که چگونه اعداد زیر ممکن است برای فرزند شما اعمال شود زیرا او بهترین وضعیت برای شما را میداند.
بقا توسط گروه خطر گروه انکولوژی کودکان (COG)
- گروه کم خطر: نرخ بقای 5 ساله کودکان در گروه کم خطر بالاتر از 95 درصد است.
- گروه در معرض خطر متوسط: کودکان در گروه با خطر متوسط، نرخ بقای 5 ساله در حدود 90 تا 95 درصد دارند.
- گروه پرخطر: نرخ بقای 5 ساله کودکان در گروه پرخطر حدود 50 درصد است.
درمان نوروبلاستوما
اگر کودک شما به نوروبلاستوما تشخیص داده شده باشد، تیم مراقبت از سرطان گزینههای درمانی را با شما در میان میگذارد. مهم است که هم فواید هر گزینه درمانی و هم خطرات و عوارض جانبی احتمالی را در نظر بگیرید.
نوروبلاستوما چگونه درمان میشود؟
چندین نوع درمان میتواند برای نوروبلاستوم استفاده شود:
جراحی نوروبلاستوما (Surgery)
از جراحی میتوان هم برای کمک به تشخیص نوروبلاستوما و هم برای درمان آن استفاده کرد. برای تومورهای کوچکتر که گسترش نیافته اند، جراحی اغلب تنها درمانی است که مورد نیاز است.
بیوپسی جراحی یا Surgical biopsy (باز یا open)
در بسیاری از موارد، پزشکان قبل از تصمیم گیری در مورد اینکه کدام درمان ممکن است بهترین کار را انجام دهد، نیاز به گرفتن نمونه ای از تومور دارند تا مطمئن شوند که بیماری نوروبلاستوما است. نمونههای تومور را میتوان در طول بیوپسی جراحی برای بررسی زیر میکروسکوپ و سایر تستهای آزمایشگاهی برداشت.
اگر تومور در شکم (belly) باشد، جراح ممکن است بیوپسی را با لاپاراسکوپ انجام دهد. این یک لوله بلند و نازک با یک دوربین فیلمبرداری کوچک در انتهای آن است. از طریق یک برش کوچک به داخل شکم گذاشته میشود تا جراح بتواند داخل آن را ببیند. سپس جراح برش کوچک دوم را ایجاد میکند تا با ابزارهای بلند و نازک به داخل شکم برسد و تکههایی از تومور را خارج کند.
جراحی برای درمان نوروبلاستوم
پس از تشخیص نوروبلاستوما، اغلب از جراحی برای حذف تا حد امکان تومور استفاده میشود. در برخی موارد، جراحی میتواند تمام (یا تقریباً تمام) تومور را حذف کند و نیازی به درمان اضافی نیست.
در طول عمل، جراح به دقت به دنبال علائم گسترش سرطان به سایر اندامها است. غدد لنفاوی مجاور (مجموعههای کوچکی از سلولهای سیستم ایمنی که اغلب سرطانها ابتدا به آنها گسترش مییابند) برداشته شده و زیر میکروسکوپ برای یافتن سلولهای سرطانی بررسی میشوند.
در صورت امکان، جراح کل تومور را بر میدارد.
اگر تومور در نزدیکی ساختارهای حیاتی باشد یا در اطراف رگهای خونی بزرگ پیچیده شده باشد، این احتمال کمتر است. حتی اگر مقداری از تومور باقی بماند، همیشه به این معنی نیست که تومور عود میکند. اینکه آیا شیمی درمانی یا سایر درمانها پس از جراحی مورد نیاز خواهند بود، بستگی به گروه خطر کودک دارد.
اگر تومور خیلی بزرگ باشد، ممکن است قبل از جراحی از شیمی درمانی برای کوچک کردن تومور و راحتتر برداشتن آن استفاده شود.
خطرات و عوارض احتمالی جراحی
خطرات ناشی از جراحی به محل تومور و وسعت عمل و همچنین سلامت کودک از قبل بستگی دارد. عوارض جدی، اگرچه نادر اند، میتواند شامل مشکلات بیهوشی، خونریزی بیش از حد، عفونتها و آسیب به عروق خونی، کلیهها یا سایر اندامها یا اعصاب باشد. اگر تومور بزرگ باشد و به عروق خونی یا اعصاب تبدیل شود، احتمال بروز عوارض بیشتر است. اکثر کودکان تا مدتی پس از عمل درد خواهند داشت اما معمولاً در صورت نیاز میتوان با داروها به این درد کمک کرد.
شیمی درمانی (Chemotherapy) برای نوروبلاستوما
شیمی درمانی (chemo) استفاده از داروهای ضد سرطان است که معمولاً در ورید تجویز میشود. این داروها وارد جریان خون میشوند و در سراسر بدن حرکت میکنند تا به سلولهای سرطانی برسند و آنها را از بین ببرند. این امر شیمی درمانی را برای درمان نوروبلاستوم مفید میکند، به خصوص اگر نتوان همه آن را با جراحی برداشت.
چه زمانی ممکن است از شیمی درمانی استفاده شود؟
اینکه آیا یک کودک مبتلا به نوروبلاستوما شیمی درمانی میکند یا نه بستگی به گروه خطر او دارد که بر اساس مرحله (وسعت) و محل سرطان، سن کودک و عوامل دیگر است.
برخی از کودکان مبتلا به نوروبلاستوما یا قبل از جراحی (شیمی درمانی نئوادجوانت یا neoadjuvant chemotherapy) یا بعد از جراحی (شیمی درمانی کمکی یا adjuvant chemotherapy) با شیمی درمانی درمان میشوند.
در موارد دیگر، به خصوص زمانی که سرطان به حدی گسترش یافته است که به طور کامل توسط جراحی برداشته نمیشود، شیمی درمانی روش درمانی اصلی است.
کدام داروهای شیمی درمانی برای نوروبلاستوما استفاده میشود؟
شیمی درمانی برای نوروبلاستوما معمولاً شامل ترکیبی از داروها است. داروهای شیمی درمانی اصلی مورد استفاده عبارتند از:
- سیکلوفسفامید (Cyclophosphamide)
- سیس پلاتین (Cisplatin) یا کربوپلاتین (carboplatin)
- وین کریستین (Vincristine)
- دوکسوروبیسین (Adriamycin)
- اتوپوزید (Etoposide)
- توپوتکان (Etoposide)
- ملفالان یا Melphalan (گاهی در طول پیوند سلولهای بنیادی استفاده میشود)
- بوسولفان یا Busulfan (گاهی در طول پیوند سلولهای بنیادی استفاده میشود)
- تیوتپا یا Thiotepa (گاهی در طول پیوند سلولهای بنیادی استفاده میشود)
رایج ترین ترکیب داروها شامل سیس پلاتین (یا کربوپلاتین)، سیکلوفسفامید، دوکسوروبیسین، وینکریستین و اتوپوزید است اما ممکن است از داروهای دیگر استفاده شود.
برای کودکان در گروه پرخطر، ممکن است داروهای دیگری نیز اضافه شود و برخی داروها ممکن است با دوزهای بالاتر تجویز شوند. این ممکن است با پیوند سلولهای بنیادی دنبال شود.
پزشکان شیمی درمانی را به صورت دوره ای انجام میدهند. درمان برای چند روز متوالی انجام میشود و پس از آن استراحت داده میشود تا بدن زمان لازم برای بهبودی داشته باشد. چرخهها معمولاً هر 3 یا 4 هفته تکرار میشوند. طول کل درمان بستگی به این دارد که کودک در کدام گروه خطر قرار دارد – گروههای پرخطر معمولاً به درمان طولانی تری نیاز دارند.
عوارض جانبی احتمالی شیمی درمانی
داروهای شیمی درمانی میتوانند سایر سلولهای بدن را که به سرعت در حال تقسیم هستند، تحت تاثیر قرار دهند که میتواند منجر به عوارض جانبی شود. عوارض جانبی شیمی درمانی به نوع و دوز داروهای داده شده و مدت زمان مصرف آنها بستگی دارد.
عوارض عمومی شیمی درمانی
عوارض جانبی رایج در بسیاری از داروهای شیمی درمانی عبارتند از:
- ریزش مو
- زخمهای دهانی
- از دست دادن اشتها
- تهوع و استفراغ
- اسهال یا یبوست
شیمی درمانی میتواند به مغز استخوان – جایی که سلولهای خونی جدید ساخته میشوند – آسیب برساند. این میتواند منجر به کاهش تعداد سلولهای خونی شود که میتواند منجر به موارد زیر شود:
- افزایش احتمال عفونت (به دلیل داشتن گلبولهای سفید بسیار کم)
- کبودی یا خونریزی آسان (به دلیل داشتن تعداد بسیار کم پلاکت خون)
- خستگی (به دلیل داشتن گلبولهای قرمز بسیار کم)
بیشتر این عوارض پس از پایان دوره درمان از بین میروند. اغلب راههایی برای کاهش این عوارض وجود دارد. به عنوان مثال، میتوان داروهایی برای کمک به پیشگیری یا کاهش تهوع و استفراغ تجویز کرد. حتماً از پزشک یا پرستار فرزندتان در مورد داروهایی که به کاهش عوارض جانبی کمک میکنند، بپرسید و به آنها اطلاع دهید که آیا کودک شما عوارض جانبی دارد تا بتوان آنها را مدیریت کرد.
عوارض جانبی برخی داروهای شیمی درمانی
در کنار اثرات ذکر شده در بالا، برخی از داروها میتوانند عوارض جانبی خاصی داشته باشند. مثلا:
سیکلوفسفامید میتواند به مثانه آسیب برساند که میتواند باعث ایجاد خون در ادرار شود. این خطر را میتوان با دادن مایعات فراوان و دارویی به نام مسنا (mesna) که به محافظت از مثانه کمک میکند، کاهش داد. این دارو همچنین میتواند به تخمدانها یا بیضهها آسیب برساند که میتواند بر باروری (توانایی بچه دار شدن) تأثیر بگذارد.
دوکسوروبیسین میتواند به قلب آسیب برساند. پزشکان سعی میکنند با محدود کردن دوزهای دوکسوروبیسین و بررسی قلب با اکوکاردیوگرام (سونوگرافی قلب) در طول درمان، این خطر را تا حد امکان کاهش دهند. این دارو همچنین در صورت نشت از ورید در حین مصرف میتواند باعث آسیب پوست شود.
سیس پلاتین و کربوپلاتین میتوانند بر کلیهها تأثیر بگذارند. دادن مایعات زیاد میتواند به کاهش این خطر کمک کند. این داروها میتوانند شنوایی را نیز تحت تأثیر قرار دهند. پزشک کودک شما ممکن است آزمایشهای شنوایی (آدیوگرافی یا audiograms) را در طول درمان یا بعد از آن تجویز کند.
وین کریستین و اتوپوزید میتوانند باعث آسیب عصبی (نوروپاتی یا neuropathy) شوند. برخی از بیماران ممکن است سوزن سوزن شدن، بی حسی، ضعف یا درد به خصوص در دستها و پاها داشته باشند.
شیمی درمانی همچنین میتواند برخی عوارض جانبی طولانی مدت داشته باشد. به عنوان مثال، برخی داروها میتوانند خطر ابتلا به نوع دیگری از سرطان (مانند لوسمی یا leukemia) را افزایش دهند. در حالی که این یک خطر جدی است، رایج نیست و افزایش اندک این خطر باید با اهمیت شیمی درمانی در درمان نوروبلاستوم سنجیده شود. برای اطلاعات بیشتر در مورد اثرات احتمالی طولانی مدت درمان، به مبحث اثرات دیر هنگام و طولانی مدت نوروبلاستوما و درمان آن مراجعه کنید.
پرتو درمانی (Radiation Therapy) برای نوروبلاستوما
پرتو درمانی از پرتوها یا ذرات پر انرژی برای از بین بردن سلولهای سرطانی استفاده میکند.
چه زمانی میتوان از پرتو درمانی استفاده کرد؟
گاهی اوقات پرتو درمانی بخشی ضروری از درمان است اما به دلیل عوارض جانبی احتمالی طولانی مدت در کودکان خردسال، پزشکان در صورت امکان از استفاده از آن اجتناب میکنند.
اکثر کودکان مبتلا به نوروبلاستوما نیازی به پرتو درمانی ندارند. معمولاً در کودکان مبتلا به نوروبلاستوما پرخطر، این روش پس از پیوند سلولهای بنیادی استفاده میشود. همچنین ممکن است برای کودکان مبتلا به نوروبلاستوما کم خطر و متوسط استفاده شود، اگر کودک علائم تهدید کننده زندگی و نیاز به درمان اضطراری برای کوچک کردن تومور داشته باشد.
پرتو درمانی چگونه انجام میشود؟
برای درمان کودکان مبتلا به نوروبلاستوما میتوان از دو نوع پرتو درمانی استفاده کرد:
- پرتو درمانی خارجی
- رادیوتراپی MIBG
پرتو درمانی خارجی (External beam radiation therapy)
پرتو درمانی خارجی از دستگاهی برای متمرکز کردن پرتوی تشعشعی بر روی سرطان از منبع تشعشع خارج از بدن استفاده میکند. این نوع درمان ممکن است در شرایط زیر مورد استفاده قرار گیرد:
- تلاش برای کوچک کردن تومورها قبل از عمل جراحی و برداشتن آسانتر آنها
- برای درمان تومورهای بزرگتر که باعث مشکلات جدی (مانند مشکلات تنفسی) میشوند و به سرعت به شیمی درمانی پاسخ نمیدهند.
- به عنوان بخشی از رژیم درمانی پس از پیوند سلولهای بنیادی در کودکان مبتلا به نوروبلاستوما پرخطر برای از بین بردن سلولهای نوروبلاستوم باقی مانده. تابش ممکن است به ناحیه تومور اولیه و سایر نواحی بدن که ممکن است دارای بیماری فعال در اسکن MIBG باشند، داده شود.
- برای کمک به تسکین درد ناشی از نوروبلاستوم پیشرفته
هنگامی که تابش تمام بدن را هدف قرار میدهد، به عنوان تابش کل بدن (total body irradiation یا TBI) شناخته میشود. این در گذشته برای کودکان مبتلا به نوروبلاستوما پرخطر قبل از پیوند سلولهای بنیادی استفاده میشد اما در حال حاضر پرتو دهی فقط پس از پیوند سلولهای بنیادی و فقط به محل تومور اولیه و هر ناحیه دیگری از بدن رایجتر است که ممکن است سلولهای نوروبلاستوما فعال داشته باشند.
قبل از شروع پرتو درمانی، تیم پرتو درمانی با آزمایشهای تصویر برداری مانند اسکنهای MRI اندازه گیریهای دقیقی را انجام میدهد تا زوایای صحیح برای نشانه گیری پرتوها و دوز مناسب تابش را تعیین کند.
همچنین ممکن است برای کودک شما یک قالب پلاستیکی شبیه قالب بدن تعبیه شود تا در طول هر جلسه درمانی در همان موقعیت قرار گیرد تا پرتو را با دقت بیشتری هدف گیری کند.
تعداد پرتو درمانیهای انجام شده بستگی به شرایط دارد.
در هر جلسه درمانی، کودک شما روی یک میز مخصوص دراز میکشد در حالی که یک دستگاه تابش را از زاویه دقیق ارسال میکند. پرتو درمانی بسیار شبیه عکس برداری با اشعه ایکس است اما دوز پرتو بسیار بالاتر است. روند درمان دردناک نیست. هر جلسه درمانی واقعی فقط چند دقیقه طول میکشد اما زمان تنظیم – قرار دادن کودک شما در موقعیت مناسب برای دریافت درمان – معمولاً بیشتر طول میکشد. ممکن است به کودکان خردسال دارو داده شود تا بخوابند تا در طول درمان حرکت نکنند.
عوارض جانبی احتمالی پرتو درمانی خارجی
پرتو درمانی گاهی اوقات بخش مهمی از درمان است اما بدن کودکان خردسال نسبت به آن بسیار حساس است، بنابراین پزشکان سعی میکنند تا حد امکان از پرتو درمانی کمتری برای جلوگیری یا محدود کردن هرگونه مشکل استفاده کنند. تابش میتواند عوارض جانبی کوتاه مدت و بلند مدت ایجاد کند که بستگی به دوز پرتو و محل هدف آن دارد.
اثرات کوتاه مدت
- اشعه میتواند بر روی پوست ناحیه تحت درمان تاثیر بگذارد. اثرات آن میتواند از تغییرات خفیف مانند آفتاب سوختگی و ریزش مو تا واکنشهای پوستی شدیدتر باشد.
- تابش اشعه به شکم (belly) میتواند باعث تهوع یا اسهال شود.
- پرتو درمانی میتواند کودک را به خصوص در أواخر روند درمان خسته کند.
پرتو درمانی همچنین میتواند عوارض جانبی شیمی درمانی را بدتر کند. با پزشک کودک خود در مورد عوارض جانبی احتمالی صحبت کنید زیرا راههایی برای تسکین برخی از آنها وجود دارد.
اثرات بلند مدت
- پرتو درمانی میتواند رشد بافتهای طبیعی بدن (مانند استخوانها) را که پرتو میشوند، کند کند، بهویژه در کودکان کوچکتر. در گذشته این منجر به مشکلاتی مانند استخوانهای کوتاه یا انحنای ستون فقرات میشد اما با دوزهای کمتر تشعشع امروزی این احتمال کمتر است.
- اشعه میتواند غده تیروئید در گردن را تحت تاثیر قرار دهد و باعث شود هورمون تیروئید کمتری تولید کند (کم کاری تیروئید). علائم کم کاری تیروئید میتواند بسیار متفاوت باشد. در کودکان، کم کاری تیروئید میتواند بر رشد و تکامل تأثیر بگذارد.
داروهای جایگزین تیروئید معمولاً تنها چیزی است که برای مدیریت کم کاری تیروئید لازم است.
- تشعشعاتی که به ناحیه قفسه سینه میرسد میتواند بر قلب و ریهها تأثیر بگذارد. این معمولاً فوراً مشکلی ایجاد نمیکند اما در برخی از کودکان ممکن است در نهایت با افزایش سن منجر به مشکلات قلبی یا ریوی شود.
- تابش به شکم در دختران میتواند به تخمدانها آسیب برساند. این ممکن است منجر به چرخههای قاعدگی غیر طبیعی یا مشکلاتی برای باردار شدن یا بچه دار شدن در آینده شود.
- تابش میتواند به DNA داخل سلولها آسیب برساند. در نتیجه، پرتو درمانی خطر ابتلا به سرطان دوم را در مناطقی که پرتو درمانی میشوند، معمولاً سالها پس از دریافت پرتو، کمی افزایش میدهد.
با بزرگتر شدن کودکان، پیگیری دقیق با پزشکان مهم است تا هر مشکلی در اسرع وقت پیدا و درمان شود. برای اطلاعات بیشتر در مورد اثرات احتمالی طولانی مدت درمان، به مبحث اثرات دیر هنگام و طولانی مدت نوروبلاستوما و درمان آن مراجعه کنید.
رادیوتراپی MIBG
همان طور که در تستهای نوروبلاستوما توضیح داده شد، MIBG یک ماده شیمیایی شبیه نوراپی نفرین است که توسط سلولهای عصبی سمپاتیک ساخته میشود. شکل کمی رادیواکتیو MIBG گاهی به عنوان بخشی از آزمایش تصویر برداری برای جستجوی سلولهای نوروبلاستوما در بدن به خون تزریق میشود. این روش اسکن MIBG نامیده میشود.
شکل بسیار رادیواکتیو MIBG همچنین میتواند برای درمان برخی از کودکان مبتلا به نوروبلاستوما پیشرفته، اغلب همراه با سایر درمانها استفاده شود. پس از تزریق به خون، MIBG به سلولهای نوروبلاستوما در هر نقطه از بدن میرود و تابش خود را ارسال میکند. (این نوع تشعشع فقط مسافت بسیار کوتاهی را طی میکند، بنابراین بر اکثر سلولهای سالم بدن تأثیر نمیگذارد.)
کودک باید چند روز پس از تزریق در اتاق مخصوص بیمارستان بماند تا زمانی که بیشتر اشعه از بدن وی خارج شود. بیشتر تابش از بدن در ادرار خارج میشود، بنابراین کودکان کوچکتر ممکن است نیاز به کاتتر در مثانه داشته باشند تا به خروج ادرار از بدن معمولاً برای چند روز کمک کند.
عوارض جانبی احتمالی
بیشتر پرتوهای ناشی از درمان با MIBG در ناحیه نوروبلاستوما باقی میماند، بنابراین اکثر کودکان عوارض جانبی جدی از این درمان ندارند. درمان با MIBG گاهی اوقات میتواند باعث تهوع و استفراغ خفیف شود. همچنین میتواند باعث شود برخی از کودکان احساس خستگی یا تنبلی کنند. در برخی از کودکان ممکن است گونهها در اثر درمان MIBG متورم شوند زیرا میتواند غدد بزاقی را تحت تاثیر قرار دهد. به ندرت ممکن است برای مدت کوتاهی باعث فشار خون بالا شود.
شیمی درمانی با دوز بالا (High-dose Chemotherapy) و پیوند سلولهای بنیادی (Stem Cell Transplant) برای نوروبلاستوما
این نوع درمان دوزهای بالاتر شیمی درمانی (chemo) را با پیوند ترکیب میکند تا سلولهای بنیادی مغز استخوان آسیب دیده توسط شیمی درمانی را جایگزین کند. اغلب در کودکان مبتلا به نوروبلاستوما پرخطر که بعید است با روشهای درمانی دیگر درمان شوند، استفاده میشود.
انجام دوزهای بالاتر شیمی درمانی ممکن است در درمان این سرطانها موثرتر باشد اما به طور معمول نمیتوان این کار را انجام داد زیرا باعث آسیب شدید به سلولهای بنیادی در مغز استخوان میشود که سلولهای خونی جدید میسازند. این میتواند منجر به کمبود سلولهای خونی شود.
پیوند سلولهای بنیادی (SCT) گاهی اوقات میتواند به پزشکان کمک کند تا با دادن شیمی درمانی با دوز بالا، این مشکل را برطرف کنند، سپس با دادن سلولهای بنیادی جدید، سلولهای مغز استخوان بیمار را جایگزین کنند.
قبل از پیوند سلولهای بنیادی، معمولاً به کودک حدود 5 ماه شیمی درمانی شدید و گاهی اوقات جراحی برای برداشتن تومور نیز داده میشود. برخی از کودکان ممکن است 2 پیوند سلولهای بنیادی را با فاصله چند ماه انجام دهند که به آن پیوند سلولهای بنیادی پشت سر هم (tandem stem cell transplants) گفته میشود.
SCT یک درمان پیچیده است که میتواند عوارض جانبی تهدید کننده زندگی ایجاد کند. اگر پزشکان فکر میکنند که فرزند شما میتواند از پیوند بهره مند شود، بهترین مکان برای انجام این کار در یک مرکز سرطان است که کارکنان آن در مورد این عمل و مدیریت دوره بهبودی تجربه دارند.
جمع آوری سلولهای بنیادی قبل از پیوند
برای اکثر کودکان مبتلا به نوروبلاستوما، سلولهای بنیادی خودشان جمع آوری شده و برای پیوند استفاده میشود.
برای کمک به آماده شدن برای جمع آوری سلولهای بنیادی، پزشکان دارویی به نام G-CSF (filgrastim) را به بیمار میدهند که به مغز استخوان کمک میکند تا سلولهای بنیادی بیشتری بسازد و به این سلولها کمک میکند تا به جریان خون حرکت کنند.
G-CSF معمولاً در پایان یک چرخه منظم شیمی درمانی شروع میشود و روزانه تجویز میشود. هنگامی که بخشی از تعداد گلبولهای سفید خون (که به عنوان تعداد مطلق نوتروفیل یا ANC شناخته میشود) به سطح معینی رسید، دوز G-CSF افزایش مییابد تا زمانی که سلولهای بنیادی کافی برای جمع آوری وجود داشته باشد.
نوع خاصی از کاتتر ورید مرکزی در بدن کودک در جای خود قرار داده میشود تا سلولهای بنیادی را بتوان طی فرآیندی به نام آفرزیس (apheresis) جمع آوری کرد. فرآیند جمع آوری خون شبیه اهدای خون است اما خون به جای رفتن به کیسه جمع آوری، به دستگاه مخصوصی میرود که سلولهای بنیادی را فیلتر میکند و سایر قسمتهای خون را به بدن کودک برمیگرداند. آفرزیس ممکن است چند ساعت طول بکشد و احتمالاً کودک شما نیاز دارد که در طول عمل صاف دراز بکشد و ثابت نگه داشته شود. این روند ممکن است طی چند روز تکرار شود. سپس سلولهای بنیادی جمع آوری شده تا زمان پیوند منجمد میشوند.
روش شیمی درمانی و پیوند با دوز بالا
به طور معمول، کودک یک روز قبل از شروع شیمی درمانی با دوز بالا در بخش SCT بیمارستان بستری میشود. آنها معمولاً تا پس از انجام شیمی درمانی و پیوند سلولهای بنیادی و تا زمانی که سلولهای بنیادی دوباره شروع به ساخت سلولهای خونی جدید کنند (معمولاً حداقل چند هفته) در بیمارستان میمانند.
کودک ابتدا شیمی درمانی با دوز بالا دریافت میکند. این کار سلولهای سرطانی بدن و همچنین سلولهای طبیعی مغز استخوان را از بین میبرد. پس از شیمی درمانی، سلولهای بنیادی منجمد ذوب شده و به عنوان تزریق خون داده میشوند. سلولهای بنیادی از طریق جریان خون حرکت کرده و در مغز استخوان کودک مستقر میشوند.
معمولاً در عرض چند هفته، سلولهای بنیادی شروع به ساخت گلبولهای سفید جدید میکنند. به زودی پس از آن، آنها شروع به ساخت گلبولهای قرمز خون و پلاکتهای جدید خواهند کرد.
تا زمانی که سلولهای خونی جدید ساخته شوند، کودک به دلیل تعداد کم گلبولهای سفید خون و همچنین خونریزی به دلیل تعداد کم پلاکتها در معرض خطر عفونت قرار دارد. برای کمک به کاهش خطر عفونت، کودک در یک اتاق بیمارستانی ویژه میماند و بازدید کنندگان باید لباس محافظ بپوشند. انتقال خون و پلاکت و درمان با آنتی بیوتیکهای IV نیز ممکن است برای کمک به پیشگیری یا درمان عفونتها یا مشکلات خونریزی استفاده شود.
کودک معمولاً تا زمانی که ANC به سطح ایمن برسد در اتاق بیمارستان میماند. سپس کودک تقریباً هر روز به مدت چندین هفته در یک کلینیک سرپایی ویزیت میشود. از آن جایی که اغلب طول میکشد تا تعداد پلاکتها به سطح ایمن برگردد، کودک ممکن است به صورت سرپایی تزریق پلاکت شود. بیماران ممکن است نیاز به مراجعه منظم به کلینیک سرپایی برای حدود 6 ماه داشته باشند، پس از این مدت پزشکان معمولی ممکن است مراقبت خود را ادامه دهند.
عوارض جانبی احتمالی
پیوند سلولهای بنیادی میتواند عوارض جانبی کوتاه مدت و طولانی مدت داشته باشد.
عوارض جانبی زودرس یا کوتاه مدت
عوارض اولیه و عوارض جانبی معمولاً ناشی از شیمی درمانی با دوز بالا است و میتواند شدید باشد. آنها در نتیجه آسیب به مغز استخوان و سایر بافتهای بدن که به سرعت در حال رشد هستند، ایجاد میشوند و میتوانند شامل موارد زیر باشند:
- تعداد کم سلولهای خونی (با خستگی و افزایش خطر عفونت و خونریزی)
- تهوع و استفراغ
- از دست دادن اشتها
- زخمهای دهانی
- اسهال
- ریزش مو
- مشکلات کبدی
یکی از شایع ترین و جدی ترین عوارض کوتاه مدت افزایش خطر ابتلا به عفونتهای جدی است. برای جلوگیری از این امر اغلب آنتی بیوتیک تجویز میشود. سایر عوارض جانبی، مانند کاهش تعداد گلبولهای قرمز و پلاکتها، ممکن است نیاز به تزریق فرآوردههای خونی یا درمانهای دیگر داشته باشد.
عوارض جانبی دیرهنگام یا طولانی مدت
برخی از عوارض و عوارض جانبی ممکن است برای مدت طولانی ادامه داشته باشند یا ممکن است تا ماهها یا سالها پس از پیوند رخ ندهند. این موارد میتواند شامل موارد زیر باشد:
- آسیب به قلب یا ریهها
- مشکلات تیروئید یا سایر غدد هورمون ساز
- مشکلات باروری
- آسیب به استخوانها یا مشکلات رشد استخوان
- ایجاد سرطان دیگری (از جمله سرطان خون) در سالها بعد
حتماً قبل از پیوند با پزشک کودک خود صحبت کنید تا در مورد اثرات بلند مدت احتمالی فرزندتان مطلع شوید. برای اطلاعات بیشتر در مورد اثرات طولانی مدت احتمالی این درمان و سایر درمانها، به مبحث اثرات دیرهنگام و طولانی مدت نوروبلاستوما و درمان آن مراجعه کنید.
درمان رتینوئید (Retinoid Therapy) برای نوروبلاستوما
رتینوئیدها مواد شیمیایی مرتبط با ویتامین A هستند. آنها به عنوان عوامل متمایز کننده (differentiating agents) شناخته میشوند زیرا تصور میشود به بالغ شدن (متمایز شدن) برخی سلولهای سرطانی به سلولهای طبیعی کمک میکنند.
در کودکان مبتلا به نوروبلاستوما پرخطر، درمان با رتینوئیدی به نام اسید 13-سیس-رتینوئیک (ایزوترتینوئین یا isotretinoin) خطر عود سرطان پس از شیمی درمانی با دوز بالا و پیوند سلولهای بنیادی را کاهش میدهد. اکثر پزشکان در حال حاضر 6 ماه پس از پیوند 13-سیس-رتینوئیک اسید را توصیه میکنند. این دارو به صورت کپسول دو بار در روز به مدت 2 هفته و سپس 2 هفته استراحت مصرف میشود.
اکنون محققان در تلاشند تا رتینوئیدهای موثرتری تولید کرده و نقش دقیق این رویکرد را در درمان نوروبلاستوما مشخص کنند.
عوارض جانبی احتمالی
شایع ترین عارضه جانبی 13-سیس-رتینوئیک اسید، خشکی و ترک خوردن لبها است. خشکی پوست یا چشم، خونریزی بینی، دردهای عضلانی و مفاصل و تغییر در ناخنها نیز ممکن است.
ایمونوتراپی (Immunotherapy) برای نوروبلاستوما
ایمونوتراپی استفاده از داروها برای کمک به سیستم ایمنی بدن خود بیمار است که سلولهای سرطانی را به طور موثرتر شناسایی و از بین ببرد. در حال حاضر چندین نوع ایمونوتراپی برای استفاده در برابر نوروبلاستوما مورد مطالعه قرار گرفته است و برخی در حال حاضر برای درمان آن استفاده میشوند.
آنتی بادیهای مونوکلونال ضد GD2 (Anti-GD2 monoclonal antibodies)
آنتی بادیهای مونوکلونال نسخههای آزمایشگاهی پروتئینهای سیستم ایمنی هستند که میتوانند به یک هدف بسیار خاص روی سلولهای بدن بچسبند. این آنتی بادیها را میتوان به خون تزریق کرد تا سلولهای سرطانی را جستجو کرده و به آنها بچسبد.
بسیاری از سلولهای نوروبلاستوما دارای مقادیر زیادی از ماده ای به نام GD2 بر روی سطوح خود هستند. آنتی بادیهای مونوکلونال که به GD2 متصل میشوند میتوانند به سیستم ایمنی بدن کمک کنند تا این سلولهای سرطانی را پیدا کرده و از بین ببرد.
دینوتوکسیماب (Unituxin یا Dinutuximab)
این آنتی بادی مونوکلونال معمولاً همراه با سایتوکاینها (cytokines، هورمونهای سیستم ایمنی) مانند GM-CSF و اینترلوکین-2 (IL-2) و همچنین ایزوترتینوئین داده میشود تا به سیستم ایمنی بدن کمک کند تا سلولهای نوروبلاستوما را شناسایی و از بین ببرد. این معمولاً به عنوان بخشی از درمان برای کودکان مبتلا به نوروبلاستوما پرخطر، پس از پیوند سلولهای بنیادی استفاده میشود.
این دارو به صورت انفوزیون داخل ورید (IV) در طی چند ساعت و به مدت 4 روز متوالی تجویز میشود. این کار تقریباً یک بار در ماه، معمولاً در مجموع برای حدود 5 سیکل درمانی انجام میشود. داروهای دیگر قبل و در طول هر انفوزیون (تزریق) برای کمک به عوارض جانبی احتمالی مانند درد یا واکنشهای انفوزیونی تجویز میشوند.
عوارض جانبی احتمالی
Dinutuximab میتواند عوارض جانبی ایجاد کند که برخی از آنها میتوانند جدی باشند. عوارض جانبی احتمالی عبارتند از:
- درد عصبی (که گاهی اوقات میتواند شدید باشد)
- نشت مایع از رگهای خونی کوچک (که میتواند منجر به فشار خون پایین، ضربان قلب سریع، تنگی نفس و تورم شود)
- واکنشهای انفوزیونی (که میتواند منجر به تورم راه هوایی، مشکل در تنفس و فشار خون پایین شود)
- مشکلات چشمی و بینایی
- تب
- استفراغ
- اسهال
- خارش
- مشکل در ادرار کردن
- عفونتها
- تعداد کم سلولهای خونی
- تغییرات در سطوح مواد معدنی در خون
سایر عوارض جانبی نیز ممکن است. با تیم درمان کودک خود صحبت کنید تا در مورد عوارض جانبی احتمالی و کارهایی که میتوان در مورد آنها انجام داد، بیشتر اطلاعات بدست آورید.
ناکسیتاماب (Naxitamab یا Danyelza)
این آنتی بادی مونوکلونال همراه با سایتوکاین (هورمون سیستم ایمنی) GM-CSF داده میشود تا به سیستم ایمنی بدن کمک کند تا سلولهای نوروبلاستوما را شناسایی کرده و از بین ببرد.
Naxitamab را میتوان در بیمارانی استفاده کرد که حداقل یک سال دارند و دارای نوروبلاستوم پرخطری هستند که در استخوان یا مغز استخوان آنها وجود دارد و پس از پاسخ اولیه به درمان دوباره برگشته یا شروع به رشد کرده است.
این دارو به صورت انفوزیون داخل ورید (IV) طی 30 تا 60 دقیقه در روزهای 1، 3 و 5 از هر چرخه 4 هفته ای تجویز میشود. داروهای دیگر قبل و در طول هر انفوزیون برای کمک به عوارض جانبی احتمالی مانند درد یا واکنشهای انفوزیونی تجویز میشوند.
عوارض جانبی احتمالی
Naxitamab میتواند عوارض جانبی ایجاد کند که برخی از آنها میتواند جدی باشد. عوارض جانبی احتمالی عبارتند از:
- درد عصبی (که گاهی اوقات میتواند شدید باشد)
- واکنشهای انفوزیونی (که میتواند منجر به تورم راه هوایی، مشکل در تنفس و فشار خون پایین شود)
- مشکلات چشمی و بینایی
- ضربان قلب سریع
- تب
- استفراغ
- سرفه
- حالت تهوع
- اسهال
- فشار خون پایین
- خارش
- مشکل در ادرار کردن
- عفونتها
- تعداد کم سلولهای خونی
- تغییرات در سطوح مواد معدنی در خون
سایر عوارض جانبی نیز ممکن است. با تیم درمان کودک خود صحبت کنید تا در مورد عوارض جانبی احتمالی و کارهایی که میتوان در مورد آنها انجام داد، بیشتر بدانید.
درمان نوروبلاستوما بر اساس گروه خطر
درمان نوروبلاستوما عمدتاً بر اساس این است که کودک در کدام گروه خطر قرار دارد. به طور کلی، کودکان کوچکتر با تومورهای کوچکتر در گروههای کم خطر قرار دارند، در حالی که کودکان بزرگتر، کودکان مبتلا به تومورهایی که در سراسر بدن پخش شده اند و کودکانی که تومورهای آنها ویژگیهای نامطلوبی دارد یا نسخههای اضافی از ژن MYCN، در گروه پرخطر قرار دارند. برخی از نوزادان مبتلا به نوروبلاستوما که در سراسر بدن پخش شده اند را میتوان کم خطر در نظر گرفت، به خصوص اگر تومور آنها کپی اضافی از MYCN یا سایر ویژگیهای نامطلوب نداشته باشد.
خطر کم
کودکانی که در معرض خطر کم قرار دارند معمولاً برای درمان نوروبلاستوما نیازی به درمان شدید ندارند. در واقع، برخی از کودکان (مخصوصاً نوزادان جوان با تومورهای کوچک) ممکن است اصلاً نیازی به درمان نداشته باشند زیرا برخی از این نوروبلاستوماها بالغ میشوند یا خود به خود از بین میروند.
اگر کودکی کم خطر باشد و تومور به راحتی برداشته شود، جراحی ممکن است تنها درمان مورد نیاز باشد. حتی اگر بعد از جراحی مقداری نوروبلاستوم باقی بماند، معمولاً میتوان بدون درمان بیشتر کودک را با دقت زیر نظر گرفت زیرا تومور باقی مانده اغلب بالغ میشود یا خود به خود از بین میرود.
اگر قسمت زیادی از تومور را نتوان برداشت، تومور بعد از جراحی بزرگتر شود یا اگر تومور علائم ایجاد کند، معمولاً شیمی درمانی (chemo) انجام میشود. یک رژیم شیمی درمانی رایج ترکیبی از کربوپلاتین، سیکلوفسفامید، دوکسوروبیسین و اتوپوزید است. اما ممکن است از ترکیبات دیگری استفاده شود.
برای آن تعداد معدودی از کودکان که علائم یک تومور کم خطر را دارند که نمیتوان بلافاصله با جراحی آن را درمان کرد، ممکن است ابتدا یک دوره کوتاه شیمی درمانی داده شود. به عنوان مثال، اگر تومور به طناب نخاعی فشار میآورد یا بر تنفس تأثیر میگذارد، ممکن است از شیمی درمانی برای کوچک کردن تومور برای کنترل علائم استفاده شود. اگر علائم با شیمی درمانی بهبود نیافتند، تهدید کننده زندگی باشند یا باعث فشردگی طناب نخاعی شوند، ممکن است یک دوره کوتاه پرتو درمانی استفاده شود.
نوزادان مبتلا به بیماری مرحله (MS) و بدون علامت اغلب میتوانند بدون هیچ درمانی با دقت تحت نظر باشند زیرا این سرطانها معمولاً بالغ میشوند یا خود به خود از بین میروند. اگر تومور باعث بروز مشکلاتی مانند بزرگ شدن کبد شود که میتواند برای نوزادان بسیار کم سن تهدید کننده باشد، ممکن است از شیمی درمانی با شدت کمتر برای کوچک کردن تومور استفاده شود.
اگر شیمی درمانی فوراً کبد را کوچک نکند، ممکن است از پرتو درمانی استفاده شود.
نوزادان کمتر از 6 ماه با تومورهای کوچک آدرنال (که فرض میشود نوروبلاستوم هستند) اغلب میتوانند بدون نیاز به جراحی یا سایر درمانها، با آزمایشهای تصویر برداری از نزدیک تحت نظر باشند. بسیاری از این تومورها به خودی خود بالغ میشوند یا از بین میروند اما اگر تومور به رشد خود ادامه دهد یا علائم ایجاد کند، ممکن است از جراحی یا شیمی درمانی استفاده شود.
خطر متوسط
جراحی بخش مهمی از روند درمان برای کودکان در معرض خطر متوسط است اما به ندرت به تنهایی کافی است. به کودکان معمولاً 4 تا 8 سیکل (حدود 12 تا 24 هفته) شیمی درمانی قبل یا بعد از جراحی داده میشود. داروهای شیمی درمانی مورد استفاده معمولاً شامل کربوپلاتین، سیکلوفسفامید، دوکسوروبیسین و اتوپوزید هستند. اگر ابتدا از شیمی درمانی استفاده شود، ممکن است جراحی برای برداشتن تومور باقی مانده انجام شود. پرتو درمانی معمولاً مورد نیاز نیست مگر اینکه تومور به خوبی به شیمی درمانی پاسخ ندهد یا اگر علائم تومور در کودک نیاز به درمان اورژانسی داشته باشد.
پزشکان همچنین در حال بررسی امکان پیگیری نوزادان و کودکان کم سن بدون علائم و ویژگیهای تومور مطلوب، به جای درمان با جراحی و یا شیمی درمانی هستند. در این روش، پزشکان با استفاده از آزمایشهای تصویر برداری تومور را از نزدیک تماشا میکنند تا مطمئن شوند که تومور از بین میرود یا بزرگتر نمیشود. اگر تومور بزرگتر شود یا کودک علائمی داشته باشد، روند درمان با شیمی درمانی آغاز میشود. برخی از مطالعات با استفاده از این رویکرد نتایج امیدوار کننده ای را نشان داده اند و اکنون مطالعات بیشتری در حال انجام است.
کودکانی که در معرض خطر متوسط هستند و به شیمی درمانی نیاز دارند از نزدیک تحت نظر قرار میگیرند تا ببینند که چگونه پس از هر 2 سیکل (6 تا 8 هفته) پاسخ میدهند. تعداد کل چرخههایی که آنها دریافت میکنند بستگی به این دارد که شیمی درمانی چقدر تومور را کوچک میکند. پزشکان امیدوارند که درمان با شیمی درمانی بر اساس این نتایج بتواند به کودکانی که دارای تومورهایی هستند که به سرعت به درمان پاسخ میدهند، کمک کند تا کمتر شیمی درمانی کنند.
خطر بالا
کودکان در معرض خطر به درمان تهاجمی تری نیاز دارند که اغلب شامل شیمی درمانی، جراحی، پرتو درمانی، پیوند سلولهای بنیادی، ایمونوتراپی و درمان با رتینوئید است. درمان اغلب در 3 مرحله انجام میشود.
القاء (Induction): هدف از این مرحله این است که سرطان با از بین بردن یا حذف هر چه بیشتر آن به مرحله بهبودی برسد. درمان معمولاً با شیمی درمانی، با استفاده از رژیمهای متناوب از چندین دارو (در ایالات متحده، به طور معمول سیس پلاتین، اتوپوزید، وین کریستین، سیکلوفسفامید، دوکسوروبیسین و توپوتکان) با دوزهای بالاتر از آن چه برای سایر گروههای در معرض خطر استفاده میشود، شروع میشود.
پزشکان همچنین در حال مطالعه استفاده از درمانهای دیگر در این مرحله هستند، مانند داروهای هدفمند برای تومورهای دارای جهش ژن ALK و رادیوتراپی MIBG برای تومورهایی که MIBG را میگیرند.
جراحی معمولاً پس از القاء انجام میشود تا تمام تومورهایی که هنوز قابل مشاهده هستند، حذف شوند.
تثبیت (Consolidation): در این مرحله از دوره درمانی فشرده تری برای خلاص شدن از شر سلولهای سرطانی باقی مانده در بدن استفاده میشود. شیمی درمانی با دوز بالا انجام میشود و به دنبال آن یک یا دو پیوند سلولهای بنیادی انجام میشود. برخی تحقیقات نشان داده اند که پیوند دو سلول بنیادی پشت سر هم (پیوند سلولهای بنیادی پشت سر هم) ممکن است بهتر از یک پیوند سلول بنیادی باشد. این در حال حاضر در آزمایشات بالینی بیشتر مورد مطالعه قرار میگیرد.
بر اساس نتایج اسکن MIBG، اغلب پس از پیوند سلولهای بنیادی به محل تومور اولیه (حتی اگر تومور با جراحی برداشته شد) و به هر قسمت دیگری از بدن که ممکن است هنوز سرطان داشته باشد، تابش داده میشود.
نگهداری (Maintenance): هدف این مرحله از درمان، تلاش برای کاهش احتمال بازگشت سرطان است. درمان معمولاً حدود 6 ماه پس از تکمیل تثبیت انجام میشود و شامل داروی رتینوئیدی 13-cis-retinoic acid (ایزوترتینوئین) و همچنین ایمونوتراپی با آنتی بادی مونوکلونال مانند دینوتوکسیماب (یونیتوکسین) و سیتوکینهای فعال کننده سیستم ایمنی (GM-CSF و IL-2) است.
نوروبلاستوم عود کننده
اگر نوروبلاستوما پس از درمان اولیه عود کند، به عنوان عود یا بازگشت (recurrence) شناخته میشود. درمان نوروبلاستوما عود کننده به عوامل زیادی بستگی دارد، از جمله گروه خطر اولیه، محل عود سرطان و درمانهایی که استفاده شده است.
برای نوروبلاستوماهای کم خطر و متوسط که در همان ناحیه ای که شروع شده اند عود میکنند، جراحی با شیمی درمانی یا بدون آن ممکن است موثر باشد.
برای سرطانهای پرخطر یا سرطانهایی که در نقاط دورتر بدن عود میکنند، درمان معمولاً شدیدتر است و ممکن است شامل ترکیبی از شیمی درمانی، جراحی و پرتو درمانی (مانند رادیوتراپی MIBG) باشد. شیمی درمانی ممکن است شامل داروهایی باشد که در طول دوره درمان اولیه استفاده نشده اند. گزینههای دیگر ممکن است شامل درمان فشرده با شیمی درمانی با دوز بالا و به دنبال آن پیوند سلولهای بنیادی، یا درمان با آنتی بادی مونوکلونال ناکسیتاماب (Danyelza) باشد.
از آن جایی که درمان این سرطانها ممکن است سخت باشد، آزمایشهای بالینی درمانهای جدیدتر، مانند سایر آنتیبادیهای مونوکلونال، درمان با سلولهای T CAR، یا سایر داروهای ضد سرطان جدید، ممکن است گزینه معقول دیگری باشد. برای کسب اطلاعات بیشتر، به مبحث چه چیزی در تحقیقات نوروبلاستوما جدید است؟ مراجعه کنید.
چه چیزی در تحقیقات نوروبلاستوما جدید است؟
تحقیقات مهمی در مورد نوروبلاستوما در حال حاضر در بسیاری از بیمارستانهای دانشگاهی، مراکز پزشکی و سایر موسسات تحقیقاتی در سراسر جهان انجام میشود. هر سال، دانشمندان اطلاعات بیشتری در مورد عوامل ایجاد کننده این بیماری و چگونگی بهبود درمان پیدا میکنند.
ژنتیک نوروبلاستوم
اکنون محققان آزمایشهای بهتری برای جستجوی تغییرات در ژنهای سلولهای نوروبلاستوما دارند. محققان ممکن است بدانند که تغییری در یک کروموزوم خاص (رشتهای از DNA در داخل سلول که شامل ژنهای آن است) رخ داده است اما هنوز باید درباره آن ژن یا بخشی از یک ژن بیشتر بدانند. چند روش مختلف وجود دارد که ژنها در سلولهای نوروبلاستوما تغییر میکنند:
- گاهی اوقات نسخههای اضافی از همان ژن (به نام تقویت کننده یا amplification) روی یک کروموزوم وجود دارد.
- گاهی اوقات یک کروموزوم میتواند دارای قطعات گمشده DNA (به نام حذف) یا قطعات اضافی DNA (به نام افزایش یا افزودن) باشد که میتواند بر روی ژنهای کروموزوم تأثیر بگذارد.
- گاهی اوقات ژنها دارای جهش (تغییرات در DNA) یا تغییرات دیگری هستند که میتوانند ژن را روشن یا خاموش کنند که میتواند بر نحوه رشد و تقسیم سلول تأثیر بگذارد.
درک تغییرات ژنی در نوروبلاستوم به محققان کمک میکند تا بفهمند کدام نوروبلاستوما احتمالاً با درمان شدیدتر درمان میشود و کدام یک به درمان تهاجمی تری نیاز دارد. برخی از این تغییرات ژنی در حال حاضر برای کمک به تیمهای مراقبت از سرطان در تعیین مرحله نوروبلاستوما و گروه خطر کودک استفاده میشود. سایر تغییرات ژنی ممکن است به محققان کمک کند درمانهای جدیدی را پیدا کنند که بر روی انواع خاصی از سلولهای نوروبلاستوما کار میکند.
در اینجا برخی از DNA و تغییرات ژنی خاص در حال بررسی هستند:
- تغییرات DNA در بازوی کوتاه کروموزوم 6 (6p22) بیشتر در نوروبلاستوماهایی که رشد تهاجمی تری دارند، دیده میشود.
- سلولهای نوروبلاستوما در کودکان بزرگتر به احتمال زیاد تغییراتی در ژن سرکوبگر تومور ATRX دارند. تومورهایی با این تغییر ژنی تمایل به رشد آهستهتری دارند اما درمان آنها نیز سختتر است. این ممکن است توضیح دهد که چرا کودکان بزرگتر تمایل به نوروبلاستومهای پرخطر دارند (که اغلب درمان آنها سخت تر است)، در حالی که کودکان کوچکتر تمایل به نوروبلاستوم با خطر کم یا متوسط دارند (که اغلب درمان آنها آسان تر است).
- تغییرات یا داشتن بیش از یک نسخه (تقویت کننده) ژنهای ALK و MYCN ویژگیهایی هستند که برای کمک به تصمیم گیری در مورد گروه خطر کودک استفاده میشوند. برخی از داروهایی که سلولها را با تغییرات ژن ALK هدف قرار میدهند، قبلاً برای درمان انواع دیگر سرطان استفاده میشدند و اکنون برای استفاده در برابر نوروبلاستومها با تغییرات ژن ALK مورد مطالعه قرار میگیرند (به مباحث زیر مراجعه کنید). برخی از دانشمندان همچنین در حال مطالعه این موضوع هستند که چگونه تغییرات ژن ALK ممکن است با نسخههای اضافی ژن MYCN در سلولهای نوروبلاستوما مرتبط باشد.
درمان
با یافتن راههایی برای بهبود درمانهای فعلی، نرخ بقای نوروبلاستوما بهتر شده است اما نرخ بقای کودکان مبتلا به نوروبلاستومای پرخطر به خوبی کودکان مبتلا به بیماری کم خطر یا متوسط نیست.
اکثر آزمایشات بالینی نوروبلاستوما پرخطر (تومورهای تهاجمی تر و درمان سخت تر) بر یافتن بهترین ترکیب از داروهای شیمی درمانی (chemo)، رژیمهای پیوند سلولهای بنیادی، ایمونوتراپی و سایر درمانهای جدید برای تلاش برای درمان کودکان بیشتر متمرکز است. مطالعات کنونی در مورد نوروبلاستوما با خطر کم و متوسط در تلاش است تا دریابد که آیا کودکان میتوانند درمان کمتری دریافت کنند و هنوز هم همین طور هستند.
شیمی درمانی
جستجو برای یافتن بهترین ترکیبات داروهای شیمی درمانی برای درمان نوروبلاستوما ادامه دارد.
چندین داروی شیمی درمانی که قبلاً برای درمان سرطانهای دیگر استفاده میشدند، مانند توپوتکان، ایرینوتکان و تموزولوماید، اکنون در ترکیب با انواع دیگر درمانها برای استفاده در برابر نوروبلاستوما پرخطر یا نوروبلاستوما که برگشته است، مورد مطالعه قرار میگیرند.
مطالعات دیگر به دنبال این هستند که ببینند آیا برخی از کودکان مبتلا به نوروبلاستوما با خطر کم یا متوسط را میتوان با شیمی درمانی کمتر (یا حتی بدون شیمی درمانی) درمان کرد. هدف این است که همچنان همان نتایج خوب را داشته باشیم اما با عوارض جانبی کمتر ناشی از درمان.
پیوند سلولهای بنیادی
پزشکان همچنین با استفاده از ترکیبهای مختلف شیمی درمانی، پرتو درمانی، رتینوئیدها، ایمونوتراپی و سایر درمانها در تلاش هستند تا میزان موفقیت کودکان مبتلا به نوروبلاستوما تهاجمی را با شیمی درمانی با دوز بالا و پیوند سلولهای بنیادی بهبود بخشند. مثلا:
- تحقیقات اخیر نشان داده است که انجام دو پیوند سلول بنیادی (پیوند پشت سر هم) به کودکان مبتلا به نوروبلاستومای پرخطر بهتر از یک پیوند سلول بنیادی است.
- پزشکان در حال بررسی این موضوع هستند که آیا استفاده از ترکیبات مختلف داروهای شیمی درمانی، مانند بوسولفان و ملفالان، ممکن است بهتر از ترکیبات شیمیایی که در حال حاضر قبل از پیوند سلولهای بنیادی استفاده میشود، موثر باشد.
- مطالعات دیگر به دنبال این هستند که آیا استفاده از سلولهای بنیادی اهدایی از شخص دیگری (پیوند سلولهای بنیادی آلوژنیک یا allogeneic stem cell transplant)، به جای بیمار (پیوند سلولهای بنیادی اتولوگ یا autologous stem cell transplant)، ممکن است به برخی از کودکان مبتلا به تومورهای سخت درمان کمک کند.
رتینوئیدها
رتینوئیدهایی مانند 13-سیس-رتینوئیک اسید (ایزوترتینوئین) میتوانند خطر عود را پس از درمان در کودکان مبتلا به نوروبلاستوما پرخطر کاهش دهند، به ویژه هنگامی که آنها با درمانهای ایمونوتراپی خاصی تجویز میشوند. دادن 13-cis-retinoic acid در ترکیب با انواع مختلف داروهای شیمی درمانی، ایمونوتراپیهایی به نام آنتی بادیهای مونوکلونال و داروهای هدفمند در تعدادی از کارآزماییهای بالینی برای کمک به تعیین ترکیبهایی که ممکن است بهترین کار را انجام دهند، مورد مطالعه قرار گرفته است.
داروهای هدفمند
دانستن اینکه چه چیزی سلولهای نوروبلاستوما را از سلولهای طبیعی متفاوت میکند، ممکن است به رویکردهای جدیدی برای درمان این بیماری منجر شود. داروهای جدیدتری که سلولهای نوروبلاستوما را به طور خاص تری نسبت به داروهای شیمی درمانی استاندارد هدف قرار میدهند، اکنون در آزمایشات بالینی مورد مطالعه قرار میگیرند. مثلا:
در برخی از نوروبلاستومها، سلولها تغییراتی در ژن ALK دارند که به رشد آنها کمک میکند. داروهایی که سلولهایی را با تغییراتی در ژن ALK هدف قرار میدهند، مانند کریزوتینیب (Xalkori)، قبلاً در درمان برخی دیگر از انواع سرطان مفید بودند. کریزوتینیب اکنون به عنوان بخشی از درمان برای کودکان مبتلا به نوروبلاستوما پرخطر در زمانی که سلولها دارای تغییرات ژن ALK هستند، مورد مطالعه قرار میگیرد. داروهای دیگری که سلولهای دارای تغییرات ALK را هدف قرار میدهند، مانند lorlatinib (Lorbrena) و ceritinib (Zykadia) نیز در حال مطالعه هستند.
در برخی از نوروبلاستومها، سلولها دارای یک مسیر سیگنال دهی بیش فعال Aurora A کیناز هستند که به رشد سلولها کمک میکند. داروهایی که مسیر Aurora A کیناز را هدف قرار میدهند، مانند اربومین LY3295668، اکنون در آزمایشات بالینی برای استفاده در برابر نوروبلاستوما مورد مطالعه قرار میگیرند.
پزشکان همچنین تغییرات ژنی درون سلولهای نوروبلاستوما را با دقت بیشتری بررسی میکنند تا ببینند آیا سایر داروهای هدفمند ممکن است مفید باشند یا خیر. برخی از داروهای هدفمندی که در این روش مورد مطالعه قرار میگیرند عبارتند از: سورافنیب، داساتینیب، وورینوستات و افلورنیتین (DMFO).
بسیاری از داروهای هدفمند دیگر در حال حاضر برای استفاده در برابر نوروبلاستوما نیز مورد مطالعه قرار میگیرند.
ایمونوتراپی
ایمونوتراپی استفاده از داروها برای کمک به سیستم ایمنی بدن خود بیمار برای مبارزه با سرطان است. چندین نوع مختلف ایمونوتراپی در حال حاضر در حال آزمایش یا استفاده در برابر نوروبلاستوما هستند.
آنتی بادیهای مونوکلونال ضد GD2
آنتی بادیهای مونوکلونال dinutuximab (Unituxin) و naxitamab (Danyelza) که GD2 را روی سلولهای نوروبلاستوما هدف قرار میدهند، اکنون در ایالات متحده برای برخی از کودکان مبتلا به نوروبلاستومای پرخطر استفاده میشوند تا به سلولهای سیستم ایمنی کمک کنند تا سلولهای سرطانی را پیدا کرده و از بین ببرند. این آنتیبادیها معمولاً پس از پیوند سلولهای بنیادی مورد استفاده قرار میگیرند اما مطالعاتی در حال انجام است تا مشخص شود که آیا ممکن است در مراحل اولیه درمان نیز مفید باشند.
آنتی بادیهای دیگری که GD2 را هدف قرار میدهند نیز در حال مطالعه هستند. به عنوان مثال، Hu14.18K322A یک آنتی بادی اصلاح شده است که ممکن است مانند سایر آنتی بادیهای GD2 بدون برخی از عوارض جانبی عمل کند. دینوتوکسیماب بتا (Qarziba یا dinutuximab beta) که مشابه دینوتوکسیماب است، اکنون در اروپا استفاده میشود.
واکسنها
چندین واکسن سرطان نیز برای استفاده در برابر نوروبلاستوما در حال مطالعه هستند. برای این واکسنها، تزریق سلولهای نوروبلاستومای اصلاح شده یا مواد دیگر انجام میشود تا سعی شود سیستم ایمنی بدن کودک به سلولهای سرطانی حمله کند. این درمانها هنوز در مراحل اولیه آزمایشات بالینی هستند.
درمان با سلول T CAR
درمان با سلولهای T CAR یک روش جدید امیدوار کننده برای جذب سلولهای ایمنی خود به نام سلولهای T برای مبارزه با سرطان با تغییر آنها در آزمایشگاه است تا بتوانند سلولهای سرطانی را پیدا کرده و از بین ببرند. سلولهای T مورد استفاده در درمانهای سلولهای T CAR با افزودن ژنی برای گیرنده آزمایشگاهی (به نام گیرنده آنتی ژن کایمریک یا chimeric antigen receptor یا CAR) در آزمایشگاه تغییر میکنند که به آنها کمک میکند به سلولهای سرطانی خاص حمله کنند.
اکنون مطالعاتی در حال انجام است تا ببینیم آیا سلولهای T CAR که GD2 (یا سایر مواد) را روی سلولهای نوروبلاستوما هدف قرار میدهند، میتوانند در درمان این بیماری مفید باشند یا خیر. اکثر این آزمایشات بالینی بسیار اولیه هستند که در حال انجام یا هنوز در مرحله برنامه ریزی هستند.
چندین نوع دیگر از ایمونوتراپی نیز برای استفاده در برابر نوروبلاستوما در حال مطالعه هستند. بسیاری از این موارد در آزمایشات بالینی بسیار اولیه هستند. اگر در مورد این یا سایر درمانهای تحقیقاتی نوروبلاستوما سؤالی دارید، با تیم مراقبت از سرطان فرزندتان صحبت کنید.
همچنین بخوانید:
- استئوسارکوم (Osteosarcoma) چیست؟ علائم، تشخیص، پیشگیری و درمان
- سارکوم بافت نرم چیست؟ انواع، علائم، تشخیص و درمان
- سرطان تیروئید (Thyroid Cancer) چیست؟ علائم، تشخیص و درمان
- تومورهای استرومال دستگاه گوارش (Gastrointestinal Stromal Tumors) چیست؟ علائم، تشخیص و درمان
مترجم: فاطمه فریادرس
سلام من پسرم توده نوروبلاستوما فوق کلیه داشت در ۷ماهگی عمل شد و کاملا در آوردیم همه ی آزمایشات ونمونه ها منفی بود چون بدنش پاک بود شیمی درمانی نکردند احتمال بهبودی کامل یاعود دوباره چندچنده؟تشکر
با پزشک مشورت کنید
سلام دخترم سرطان نروبلاستوما داره
از ۱۳ سالگی الان ۱۹ سالشه
سه مرحله عود بیماری
و پیوند سلولهای بنیادی آنالوگ و عود
الان چیکار کنیم؟
به پزشک مراجعه کنید
جراحی گانگلیونوروما کنار ناف جهت خارج کردن او، عمل سختیه و دوره بهبودیش چقدر طول میکشه
با پزشک خود مشورت کنید
سلام
چند وقت پیش پسرمو برای عکس برداری mibg بخاطر نوروبلاستوم بردیم.بما گفتن توده سرطانی مشاهده نشده و تبدیل به غیر سرطانی شده.
ما شیمیدرمانی را همچنان ادامه دادیم و دکتر مارو فرستاد mri که در اون سایز توده خیلی کوچیکتر شده بود.
میخاستم بدونم اون توده نوروبلاستوم سرطانی هست یا غیر سرطانی؟
و اصلا ممکنه هیچ وقت از بین نره؟آخه دوره شیمی درمانی تموم شده
سلام، این سوالات نیاز به تشخیص پزشک دارد از مطالب اینترنتی بپرهیزید. این مطلب صرفا جهت اطلاعات عمومی است.
سلام
من چند وقت پیش پسرمو که مشکل نوروبلاستوم داره رو برای عکس برداری mibgبردم.نتیجه ای که به من اعلام کردن این بود که به نسبت قبل توده خیلی کوچیک شده و سرطانی نیست.
آیا ممکنه خطا داشته باشه و توده هنوز سرطانی باشه؟
آیا توده ممکنه هویت توده عوض بشه و دیگه سرطانی نباشه؟
به پزشک مراجعه کنید
ممنون از مطالب مفید و کاملتون.
ای کاش در انتها این موضوع مطرح میشد که پس از شیمی درمانی یا هر درمان اولیه ، چند درصد نیاز به درمان مجدد داشته و چند درصد درمان قطعی و بهبودی کامل رو پیدا کردند.
ممنون
خوشحالیم که براتون مفید بوده.
اطلاعات آماری معمولا متفاوت است بهتر است بروز سرچ کنید.
بسیار بسیار سپاس از اطلاعات جامعتون. با اینهمه جستجو تا حالا با اینهمه اطلاعات رو به رو نشده بودم
خوشحالیم که مفید واقع شده