



مقدمهای بر بیماری آلپرز
بیماری آلپرز (Alpers Disease) یک اختلال عصبی پیشرونده است که در دوران کودکی شروع میشود و در بسیاری از موارد با بیماری جدی کبدی پیچیده میشود. علائم بیماری آلپرز شامل افزایش تون عضلانی همراه با رفلکسهای اغراق آمیز (اسپاستیسیته یا spasticity)، تشنج و از دست دادن توانایی شناختی (زوال عقل یا dementia) است.
مترادفها
- Alpers انحطاط منتشر ماده خاکستری مغز با سیروز کبدی (Alpers Diffuse Degeneration of Cerebral Gray Matter with Hepatic Cirrhosis)
- فلج اطفال پیشرونده Alpers Infantile Poliodystrophy (Alpers Progressive Infantile Poliodystrophy)
- دژنراسیون منتشر مغزی در دوران نوزادی (Diffuse Cerebral Degeneration in Infancy)
- فلج اطفال سربری پیشرونده (Poliodystrophia Cerebri Progressiva)
- پلیودیستروفی مغزی پیشرونده (Progressive Cerebral Poliodystrophy)
علائم و نشانههای بیماری آلپرز
بیماری آلپرز معمولا در اوایل دوران کودکی شروع میشود و معمولاً با تشنج در هر سنی بین 3 ماهگی تا 5 سالگی نشان داده میشود. این بیماری با عدم هماهنگی حرکات، فلج جزئی، تشنج و انقباض عضلانی مشخص میشود. کودک قادر به دستیابی به تون ماهیچه طبیعی (هیپوتونی یا hypotonia) نیست، با این حال به نظر میرسد اندامها سفت هستند. در معاینه MRI افزایش تراکم ماده خاکستری در مغز مشاهده میشود. معمولا اما نه همیشه، بیماری آلپرز با آسیب کبدی همراه است.
عقب ماندگی ذهنی ممکن است شدید و پیشرونده باشد. از دست دادن کارکردهای فکری مانند تفکر، به خاطر سپردن و استدلال نیز ممکن است با عملکرد روزانه فرد (زوال عقل یا dementia) تداخل داشته باشد. در مراحل بعدی، بیماران ممکن است کنترل حرکت بازوها و پاهای خود را از دست بدهند (کوادری پلژی اسپاستیک یا spastic quadriplegia). کبد ممکن است سیروز شود و به طور کامل از کار بیفتد یا ممکن است فراتر از علائم زردی پیشرفت نکند. افراد مبتلا همچنین ممکن است در نتیجه آتروفی بینایی با تحلیل رفتن عصب بینایی نابینا شوند.
علل بیماری آلپرز
بسیاری از محققان بر این باورند که سندرم آلپرز، به جای اینکه یک اختلال مشخص باشد، یک موجودیت بالینی است (به عنوان مثال، دژنراسیون ماده خاکستری مغز در ارتباط با بیماری کبد) که ممکن است به دلایل مختلف باشد. در برخی موارد، اعتقاد بر این است که این سندرم ممکن است به عنوان یک صفت ژنتیکی اتوزومال مغلوب به ارث برسد. در موارد دیگر، پزشکان این اختلال را به یک مولکول پریون (prion) یا پریون مانند (prion-like) نسبت میدهند.
برخی از محققان بر این باورند که افراد خاصی ممکن است استعداد ژنتیکی برای بیماری آلپرز را به ارث ببرند. در چنین مواردی، برخی عوامل محیطی در ترکیب با چنین استعداد ژنتیکی ممکن است در نهایت منجر به بروز اختلال شود. تحقیقات همچنین نشان داده اند که برخی از نقصهای متابولیک یا ناهنجاریهای میتوکندری ممکن است نقشی در ایجاد این اختلال داشته باشند.
ویژگیهای انسانی، از جمله بیماریهای ژنتیکی کلاسیک، محصول تعامل دو ژن است که یکی از پدر و دیگری از مادر دریافت میشود. در اختلالات مغلوب، بیماری آلپرز ظاهر نمیشود مگر اینکه فرد ژن معیوب یکسانی را برای همان ویژگی از هر یک از والدین به ارث ببرد. اگر فردی یک ژن طبیعی و یک ژن برای بیماری دریافت کند، فرد ناقل بیماری خواهد بود اما معمولاً علائمی از خود نشان نخواهد داد.
خطر انتقال این بیماری به فرزندان یک زوج که هر دو ناقل اختلال مغلوب هستند، 25 درصد است. 50 درصد از فرزندان آنها در معرض خطر ناقل این بیماری هستند اما به طور کلی علائم این اختلال را نشان نمیدهند. 25 درصد از فرزندانشان ممکن است هر دو ژن طبیعی را دریافت کنند، یکی از هر والدین و از نظر ژنتیکی طبیعی باشند (برای آن ویژگی خاص). خطر برای هر بارداری یکسان است.
جمعیتهای آسیب دیده
تصور میشود که بیماری آلپرز معمولاً در دوران کودکی به تعداد مساوی مردان و زنان را تحت تأثیر قرار میدهد. با این حال، برخی از پزشکان متقاعد شده اند که دشواری تشخیص باعث میشود تخمین فراوانی این اختلال دقیق نباشد. این احتمال وجود دارد که بیماری آلپرز کمتر از یک (1) نفر در هر 200000 نفر از جمعیت را مبتلا کند.
اختلالات با علائم مشابه بیماری آلپرز
علائم اختلالات زیر میتواند مشابه علائم بیماری آلپرز باشد. مقایسه ممکن است برای تشخیص افتراقی مفید باشد:
صرع میوکلونیک (Myoclonic epilepsy) یک اختلال عصبی ارثی است که از طریق ژنهای مغلوب به ارث میرسد. با انقباضات کوتاه ناگهانی گروهی از عضلات مشخص میشود. شروع آن معمولا بین شش تا شانزده سالگی است. در دوره اولیه تشنج، از دست دادن هوشیاری وجود دارد. پس از سالها حملات با دفعات و شدت فزاینده، اسپاسمهای عضلات صورت، تنه، بازوها و پاها تشدید میشوند. بدون درمان، این نوع صرع میتواند منجر به زوال عقل پیشرونده شود.
بیماری لی (Leigh’s disease) یک اختلال متابولیک ژنتیکی است که با ضایعات مغز، نخاع، عصب بینایی و در برخی موارد، بزرگ شدن قلب مشخص میشود. این اختلال معمولاً ابتدا در دوران شیرخوارگی تشخیص داده میشود اما ممکن است بعداً شروع شود. علائم در دوران شیرخوارگی ممکن است شامل وزن کم بدن، رشد آهسته، لرزش، تغییرات پوستی و اختلال در الگوهای تنفسی باشد. اختلالات عصبی پیشرونده، عقب ماندگی ذهنی، گفتار نامفهوم و از دست دادن هماهنگی حرکتی (آتاکسی یا ataxia) ممکن است رخ دهد. ناهنجاریهای حرکت چشم و سایر مشکلات بینایی ممکن است در مواردی که دیرتر شروع میشوند ایجاد شود.
انسفالوپاتی ورنیکه (Wernicke encephalopathy) یک اختلال دژنراتیو مغزی (degenerative brain disorder) است که با کمبود تیامین مشخص میشود. با از دست دادن هماهنگی (آتاکسی) و بی تفاوتی، سردرگمی، سرگردانی یا هذیان مشخص میشود. همچنین ممکن است اختلالات بینایی مختلفی ایجاد شود. این اختلال اغلب همراه با سندرم کورساکوف (Korsakoff syndrome) رخ میدهد که شامل کمبود ویتامین B1 (تیامین) است که معمولاً ناشی از اعتیاد به الکل است. آنسفالوپاتی Wernicke میتواند به شدت ناتوان کننده و تهدید کننده زندگی باشد اگر به موقع تشخیص داده نشده و درمان نشود.
بیماری باتن (Batten disease) یک اختلال ارثی ذخیره چربی است که به صورت یک صفت مغلوب منتقل میشود. این بیماری با نارسایی بینایی پیشرونده سریع (آتروفی بینایی یا optic atrophy)، زوال عقل، تشنج، از دست دادن هماهنگی عضلانی (آتاکسی یا ataxia) و انحنای جانبی ستون فقرات به سمت عقب (کیفوسکلیوز یا kyphoscoliosis) مشخص میشود. بیماری باتن که بیشتر در خانوادههای سفید پوست از اجداد اسکاندیناویایی اروپای شمالی رخ میدهد، معمولاً بین پنج تا هفت سالگی شروع میشود.
بیماری تای ساکس (Tay-Sachs disease) یک اختلال ژنتیکی در کودکان است که باعث تخریب پیشرونده سیستم عصبی مرکزی میشود. به طور کلی در میان کودکان میراث یهودی اروپای شرقی یافت میشود. نوزادان مبتلا به بیماری تای ساکس در بدو تولد طبیعی به نظر میرسند و به نظر میرسد تا سن حدود شش ماهگی به طور طبیعی رشد میکنند.
اولین علائم بیماری متفاوت است و در سنین مختلف آشکار میشود. این علائم ممکن است شامل کند شدن رشد، از دست دادن بینایی محیطی، پاسخ غیر طبیعی مبهوت، پیشرفت مشکلات تغذیه، ضعف، بی قراری و لکههای قرمز گیلاسی روی شبکیه باشد. در سن یک سالگی، تشنجهای مکرر، از دست دادن مهارتها و هماهنگی عضلانی از قبل آموخته شده، کوری، عقب ماندگی ذهنی، شلی و یا فلج ممکن است رخ دهد. این اختلال به عنوان یک صفت مغلوب به ارث میرسد.
تشخیص بیماری آلپرز
سندرم آلپرز معمولاً در دوران نوزادی بر اساس ارزیابی بالینی کامل، شرح حال دقیق بیمار و انواع آزمایشات تخصصی تشخیص داده میشود. چنین آزمایشاتی ممکن است شامل مطالعات تصویر برداری تخصصی از مغز باشد که ممکن است انحطاط بخش خارجی (قشر مغز یا cerebral cortex) و در برخی موارد، سایر نواحی مغز را نشان دهد.
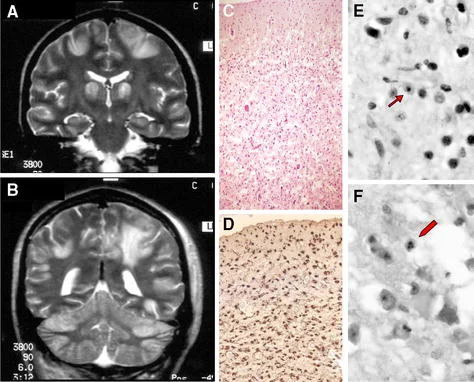
الکتروانسفالوگرافی (Electroencephalography یا EEG) که تکانههای الکتریکی مغز را ثبت میکند، ممکن است کاهش کلی فعالیت الکتریکی مغز و یا سایر ناهنجاریهای تخلیه الکتریکی مشخصه فعالیت تشنج را نشان دهد. تنها تایید پس از مرگ با بیوپسی مغز امکان پذیر است.
درمان بیماری آلپرز
هیچ درمانی در دسترس نیست که پیشرفت بیماری آلپرز را متوقف کند. با این حال، برخی از علائم را میتوان به منظور ایجاد آسایش هر چه بیشتر بیمار تحت شرایط، درمان کرد. داروهایی برای درمان دفعات تشنج، مقابله با اسپاسم عضلانی و درد مفاصل و درمان عفونت وجود دارد.
فیزیوتراپیستها ممکن است بتوانند به والدین کمک کنند تا موقعیتهای راحت تری را برای کودک در حالت نشسته یا ایستاده پیدا کنند. ماساژ اغلب استرس ناشی از آن را کاهش میدهد. تمام درمانهای سندرم آلپرز علامتی و حمایتی است.
همچنین بخوانید:
- سندرم سروتونین چیست؟ علائم، علل، درمان و پیشگیری
- سندرم آنجلمن چیست؟ ویژگیها، علل، تشخیص و درمان
- سندرم هانتر چیست؟
مترجم: فاطمه فریادرس