


بیماری ها
بیماری تای ساکس چیست؟
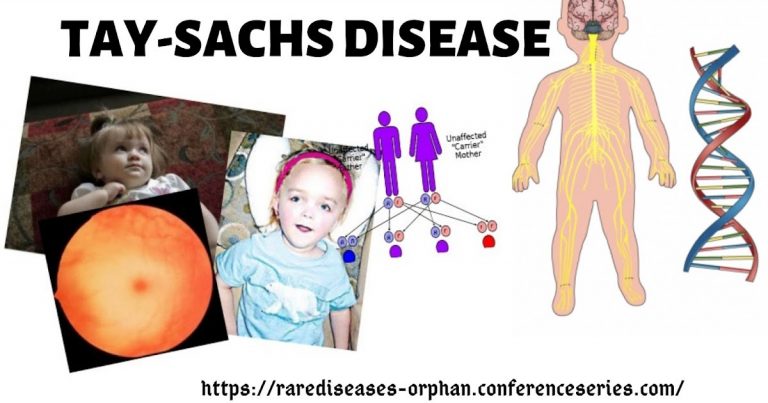
بیماری تای ساکس یک بیماری ژنتیکی است. Tay-Sachs ناشی از دریافت دو ژن معیوب HEXA توسط یک کودک از هر والدین است. علائم بیماری تای ساکس شامل عدم دستیابی به نقاط عطف حرکتی مانند نشستن و ایستادن است. نوزادانی که با تای ساکس متولد میشوند اغلب در سنین جوانی می میرند. آزمایش ژنتیک میتواند در تصمیم گیری درباره تنظیم خانواده کمک کند.
بیماری تای ساکس سلولهای عصبی مغز و نخاع را درگیر میکند. نوزادان مبتلا به Tay-Sachs فاقد آنزیم خاصی هستند که پروتئینی است که باعث واکنشهای شیمیایی در سلولها میشود. کمبود آنزیم هگزوزامینیداز A باعث جمع شدن یک ماده چرب میشود. تجمع این ماده، گانگلیوزید GM2 ، منجر به علائم Tay-Sachs مانند ضعف عضلانی میشود.
تای ساکس یک بیماری ژنتیکی است. علت آن تغییر در یک جفت ژن است که از والدین به ارث رسیده است. این یک بیماری پیشرونده است، به این معنی که با گذشت زمان بدتر میشود. کودکانی که با تای ساکس متولد میشوند اغلب تا 4 سالگی میمیرند، معمولاً به دلیل عوارض ذات الریه. هیچ درمانی وجود ندارد، زیرا درمان با هدف حمایت از کودک و راحت نگه داشتن او انجام میشود.
آزمایش ژنتیک برای زوجهایی که ممکن است با خطر بیشتری برای بچه دار شدن با Tay-Sachs مواجه شوند، در دسترس است. آزمایش و مشاوره ژنتیکی میتواند به والدین آینده در تصمیم گیری آگاهانه درباره تنظیم خانواده کمک کند.
بیماری تای ساکس به عنوان یک بیماری ذخیره سازی لیزوزومی طبقه بندی میشود. لیزوزومها واحدهای اصلی گوارش در سلولها هستند. آنزیمهای درون لیزوزومها مواد مغذی شامل کربوهیدراتها و چربیهای پیچیده (مانند گلیکوسفنگولیپیدها) را تجزیه یا “هضم” میکنند. هنگامی که یکی از این آنزیمهای لیزوزومی (مانند هگزوزامینیداز A) مفقود یا بیاثر است، گلیکوسفنگولیپیدها در لیزوزوم شروع به تجمع میکنند. اگر تجمع بیش از حد این مواد در لیزوزوم وجود داشته باشد، سلولهای سیستم عصبی تحلیل میروند و میمیرند و باعث ایجاد واکنش التهابی میشود که آسیب بافت اطراف را تقویت میکند.
شایعترین نوع بیماری تای ساکس، شکل نوزادان است، که میتواند در حدود 6 ماهگی به عنوان کاهش بینایی و پاسخ مبهم و غافلگیر کننده ظاهر شود و در نهایت به از دست دادن تدریجی مهارتها و تشنجها در سن 2 سالگی و مرگ زودرس، معمولاً در سن4 /5 سالگی. همچنین یک نسخه نوجوانان از این بیماری در حدود 5 سالگی شروع میشود و اشکال بالغ بیماری Tay-Sachs نیز به عنوان بیماری Tay Sachs دیررس شناخته میشود (LOTS) که از اواخر نوجوانی شروع میشود و فراتر. هر سه شکل بیماری تای ساکس به صورت اتوزومی مغلوب به ارث میرسد و سن شروع تابعی از مقدار، در صورت وجود، فعالیت باقیمانده آنزیم است.
علائم و نشانهها
بیماری تای ساکس نوزادان
شکل نوزادی بیماری Tay-Sachs با فقدان کامل یا تقریباً کامل فعالیت آنزیم هگزوزامینیداز A مشخص میشود. این اختلال اغلب به سرعت پیشرفت میکند و منجر به وخامت قابل توجهی از نظر شناختی و جسمی میشود.
نوزادان ممکن است در بدو تولد کاملاً سالم به نظر برسند. علائم اولیه، که معمولاً بین 3 تا 6 ماه ایجاد میشود، میتواند شامل ضعف خفیف عضلانی، لرزش یا تکان خوردن ماهیچهها (تکانهای میوکلونیک) و پاسخ مبهم و غافلگیر کننده باشد، مانند زمانی که سر و صدای ناگهانی یا غیرمنتظرهای وجود دارد. واکنش وحشتناک ممکن است تا حدی به دلیل افزایش حساسیت به صدا (حساسیت بیش از حد صوتی) باشد.
بین شش تا 10 ماهگی، نوزادان مبتلا ممکن است مهارتهای حرکتی جدیدی را به دست نیاورند. آنها ممکن است دیگر تماس چشمی برقرار نکنند و ممکن است حرکات غیر معمول چشم وجود داشته باشد. آنها ممکن است بیحال و تحریک پذیر باشند. با افزایش سن نوزادان مبتلا، ممکن است رشد آهسته، ضعف پیشرونده ماهیچهها و کاهش تون عضلانی (هیپوتونی) را تجربه کنند. نوزادان مبتلا همچنین ممکن است از دست دادن تدریجی بینایی، اسپاسم عضلانی غیر ارادی (میوکلونوس)، حرکات آرام و سفت (اسپاسم) و از دست دادن مهارتهای قبلی (به عنوان مثال، پس رفتگی حرکتی) مانند خزیدن یا نشستن را نشان دهند.
با افزایش سن نوزادان، عوارض جدیتری مانند تشنج، مشکل در بلع، از دست دادن بینایی، فلج و کم شنوایی پیشرونده ایجاد میشود. نقصهای شناختی اضافی ممکن است شامل سردرگمی، عدم تمرکز و/یا کاهش تواناییهای فکری شود. سرانجام، نوزادان ممکن است نسبت به محیط اطراف خود واکنش نشان ندهند. در سن سه تا پنج سالگی، عوارض تهدید کننده زندگی مانند پنومونی آسپیراتیو منجر به نارسایی تنفسی میشود.
نوجوانان (نوع حاد) بیماری تای ساکس
شروع بیماری نوجوانان تای ساکس میتواند بین 2 تا 10 سالگی باشد. یکی از اولین نشانهها اغلب ناشیانه و ناهماهنگ است. این امر به این دلیل رخ میدهد که کودکان مبتلا با کنترل حرکات بدن خود (آتاکسی) مشکل دارند. کودکان تمایل به از دست دادن تدریجی گفتار، مهارتهای زندگی و تواناییهای فکری دارند. افراد مبتلا ممکن است یک لکه قرمز گیلاسی در چشم ایجاد کنند یا نکنند. تخریب اعصاب بینایی (آتروفی بینایی) ممکن است رخ دهد. برخی از کودکان ممکن است مبتلا به رتینیت پیگمنتوزا باشند، از دست دادن و انحطاط پیشرونده سلولهای شبکیه که در آن ابتدا اشکال و رنگها کدگذاری میشوند. کودکان مبتلا کمتر به محیط اطراف خود پاسخ میدهند. عوارض تهدید کننده زندگی معمولاً در حدود 15 سالگی رخ میدهد.
بیماری تای ساکس دیر شروع شونده
علائم مرتبط با بیماری تای ساکس دیر شروع شونده بسیار متفاوت است. شروع بیماری ممکن است از اواخر نوجوانی تا هر زمان در بزرگسالی متفاوت باشد. این تنوع حتی ممکن است در اعضای آسیب دیده یک خانواده رخ دهد. به عنوان مثال، در یک خانواده معین ممکن است یک نفر در 20 سالگی علائم داشته باشد، در حالی که شخص دیگری با علائم جزئی نسبتاً خفیف به 60 یا 70 سالگی میرسد. این اختلال بسیار کندتر از اشکال نوزادان یا نوجوانان بیماری پیشرفت میکند.
علائم اولیه مرتبط با بیماری دیر هنگام Tay-Sachs ممکن است شامل ضعف و تحلیل رفتن پیشرونده عضلات (آتروفی نوروژنیک)، عدم هماهنگی و بیحوصلگی ناشی از اختلال عملکرد مخچه (آتاکسی) یا مشکلات حاد روانپزشکی باشد. با افزایش سن افراد دچار گرفتگی عضلات، ضعف و تحلیل رفتن ماهیچهها میشوند که به طور معمول عضلات چهارسر و خم کننده ران و بعداً عضلات سه سر را تحت تأثیر قرار میدهند. بیماران باید زانوها را در حالت هایپر اکستنشن قفل کنند تا بتوانند بایستند و وزن خود را تحمل کنند. عدم انجام این کار منجر به سقوط و در نهایت به نیاز به وسیلهای برای کمک به راه رفتن میشود. بیماران ممکن است لرزش و اختلال در گفتار پیش رونده (دیسارتریا) را نشان دهند. مشکلات بلع (دیسفاژی) ممکن است در اواخر بیماری ظاهر شود. برخی از افراد مبتلا مشکلات حاد روانپزشکی (شیدایی، افسردگی حاد یا روان پریشی) را تجربه میکنند که ممکن است نیاز به مراقبتهای روانپزشکی فوری داشته باشد. با گذشت زمان، مشکلات شناختی از جمله اختلال عملکرد اجرایی و برخی از مشکلات حافظه ممکن است ظاهر شود.
علل
بیماری تای ساکس در اثر تغییر (جهش) در ژن زیر واحد هگزوزامینیداز آلفا (HEXA) ایجاد میشود. ژنها دستورالعمل ساختار اولیه پروتئینها را ارائه میدهند که همه آنها نقش مهمی در بسیاری از عملکردها و ساختار بدن ایفا میکنند. هنگامی که جهش در یک ژن رخ میدهد، محصول پروتئینی ممکن است معیوب، ناکارآمد باشد یا وجود نداشته باشد. بسته به عملکرد پروتئین، این میتواند بر بسیاری از سیستمهای بدن از جمله مغز تأثیر بگذارد.
ژن HEXA ساختار پروتئین HEXA را که زیر واحد آنزیم هگزوزامینیداز A. است کد میکند. بیش از 80 جهش مختلف ژن HEXA در افراد مبتلا به این بیماری شناسایی شده است. به ارث بردن دو نسخه جهش یافته از ژن HEXA (هموزیگوت) باعث کمبود آنزیم هگزوزامینیداز A میشود که برای تجزیه GM2-گانگلیوزید (یک گلیکوسفنگولیپید) در سلولهای بدن ضروری است. شکست در تجزیه GM2– گانگلیوزید منجر به تجمع غیرطبیعی آن در مغز و سلولهای عصبی میشود و در نهایت منجر به وخامت پیشرونده سیستم عصبی مرکزی میشود.
بیماری تای ساکس به عنوان یک بیماری اتوزومی مغلوب به ارث میرسد. اختلالات ژنتیکی مغلوب زمانی رخ میدهد که فرد دو نسخه از یک ژن غیر طبیعی را به ارث میبرد، یعنی از هر دو والد. اگر فردی یک نسخه طبیعی از ژن را از یکی از والدین و یک نسخه غیر طبیعی (جهش یافته) از ژن را از والدین دیگر به ارث ببرد، آن فرد ناقل بیماری خواهد بود اما به این بیماری مبتلا نمیشود. خطر دو والدین ناقل برای انتقال ژن تغییر یافته و داشتن فرزند مبتلا 25٪ در هر بارداری است. خطر بچه دار شدن مانند والدین ناقل 50 درصد در هر بارداری است. شانس دریافت ژنهای طبیعی از هر والدین برای کودک 25 درصد است. خطر برای مردان و زنان یکسان است.
تشخیص
تشخیص بیماری تای ساکس ممکن است با یک ارزیابی بالینی کامل و آزمایشهای تخصصی مانند آزمایش خون که سطح فعالیت آنزیم هگزوزامینیداز A. را اندازه گیری میکند، تأیید شود. آزمایش ژنتیکی مولکولی برای جهش در ژن HEXA میتواند تشخیص بیماری تای ساکس را تایید کند. به با ظهور پانلهای ژنی گستردهتر و تعیین توالی اگزوم و کل ژنوم، بیماران بیشتری در ابتدا با آزمایش مولکولی و سپس تأیید آنزیمی تشخیص داده میشوند.
این احتمال وجود دارد که تشخیص بیماری تای ساکس قبل از تولد (قبل از تولد) بر اساس آزمایشات تخصصی مانند آمنیوسنتز و نمونه گیری از پرزهای کوریونی (CVS) مشکوک باشد. در طول آمنیوسنتز، نمونهای از مایع که جنین در حال رشد را احاطه کرده است برداشته میشود، در حالی که CVS شامل برداشتن نمونههای بافتی از قسمتی از جفت است. این نمونهها برای تعیین فعالیت هگزوزامینیداز A مورد مطالعه قرار میگیرند. عدم فعالیت یا کاهش شدید فعالیت، تشخیص را نشان میدهد. تشخیص قبل از تولد نیز از طریق آزمایش ژنتیکی مولکولی نمونههای بافتی بدست آمده از طریق CVS یا آمنیوسنتز امکان پذیر است، به ویژه اگر جهشهای ایجاد کننده بیماری در ژن HEXA در خانواده شناخته شده باشد.
آزمایش حامل بیماری Tay-Sachs میتواند از طریق نمونه خون انجام شود و مشخص میکند که آیا فردی یک نسخه از ژن HEXA عامل بیماری را حمل میکند یا خیر. بستگان افراد مبتلا به بیماری تای ساکس را میتوان برای تعیین ناقل بودن آنها آزمایش کرد.
برای زوج هایی که متوجه میشوند حامل هستند، چندین گزینه برای تشکیل خانواده وجود دارد. این گزینهها شامل فن آوریهای کمک باروری (ART) مانند لقاح آزمایشگاهی (IVF) و فرزندخواندگی است. از زوجین خواسته میشود تا در مورد این گزینهها با مشاور ژنتیک مشورت کنند.
درمانهای استاندارد
هیچ درمان مورد تاییدی برای بیماری تای ساکس وجود ندارد. درمان به سمت مدیریت علائم فردی هدایت میشود. درمان ممکن است مستلزم تلاش هماهنگ گروهی از متخصصان باشد. متخصصان اطفال، متخصصان مغز و اعصاب، آسیب شناسان گفتار، متخصصانی که مشکلات شنوایی را ارزیابی و درمان میکنند (شنوایی شناسان)، متخصصان چشم و سایر متخصصان مراقبتهای بهداشتی ممکن است برای تدوین برنامهای برای درمان کودک مبتلا نیاز به همکاری داشته باشند.
به دلیل مشکلات تغذیهای، نوزادان باید از نظر وضعیت تغذیه و هیدراتاسیون مناسب تحت نظر باشند. ممکن است حمایت و مکمل تغذیهای لازم باشد. گاهی اوقات، قرار دادن لوله تغذیه برای جلوگیری از ورود تصادفی غذا، مایع یا سایر مواد خارجی به ریهها (آسپیراسیون) ضروری است.
داروهای ضد تشنج ممکن است برای درمان تشنج در برخی از افراد مبتلا به بیماری تای ساکس استفاده شود، اما ممکن است در همه افراد موثر نباشد. انواع و دفعات تشنج در برخی افراد ممکن است با گذشت زمان تغییر کند که نیاز به تغییر در نوع دارو یا دوز آن دارد.
مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها توصیه میشود. حمایت روانی اجتماعی برای تمام اعضای خانواده توصیه میشود.
درمان جایگزینی آنزیمی (ERT) برای اختلالات ذخیره لیزوزومی در نظر گرفته و توسعه یافته است. در بیماری تای ساکس، درمان جایگزینی آنزیم شامل جایگزینی هگزوزامینیدات A. مفقود شده است. ناتوانی در یافتن راهی برای انتقال آنزیم جایگزین (یک مولکول بزرگ) برای عبور از سد خونی مغزی این استراتژی را محدود میکند.
درمان چاپرون نیز برای درمان بیماری تای ساکس مورد مطالعه قرار میگیرد. این نوع درمان شامل مولکولهای بسیار کوچکی به نام چاپرون است که به آنزیمهای هگزوزامینیداز A تازه سنتز شده متصل میشوند. این رویکرد به دنبال محافظت از تخریب هگزوزامینیداز A غیرطبیعی در داخل سلول ها قبل از تخریب است، بنابراین اجازه میدهد هگزوزامینیداز A، هر چند کم، فعالیت خود را برای مدت زمان طولانیتری اعمال کند. چاپرونی به نام پیریمتامین به عنوان درمانی برای بیماری تای ساکس مورد مطالعه قرار گرفته است. افراد مبتلا که دارو مصرف میکردند، افزایش فعالیت هگزوزامینیداز A را نشان دادند. با این حال، افزایش فعالیت منجر به بهبود قابل توجه علائم نشد و با عوارض جانبی نامطلوب همراه بود.
ژن درمانی نیز به عنوان رویکرد احتمالی دیگری برای درمان برخی از اختلالات ذخیره لیزوزومی مورد مطالعه قرار میگیرد. در ژن درمانی، ژن معیوب موجود در بیمار با یک ژن معمولی جایگزین میشود تا تولید آنزیم فعال را فعال کرده و از پیشرفت بیماری جلوگیری کند. با توجه به انتقال دائمی ژن طبیعی، که میتواند آنزیم فعال را در همه نقاط بیماری تولید کند، از نظر تئوریک این نوع درمان به احتمال زیاد منجر به “بهبود” میشود. کنسرسیوم Tay-Sachs Gene Therapy (TSGT) گروهی مشترک از دانشمندان است که به دنبال ترجمه اطلاعات حاصل از مطالعات مدل حیوانی به آزمایشات بالینی هستند.
مطالعه صدها مطلب علمی در حوزه بیولوژی
آرشیو جدیدترین خبرهای روز دنیای بیولوژی
مترجم: غزل زارعی