


دستهبندی نشده
تشخیص واریانت های ساختاری CNV ها
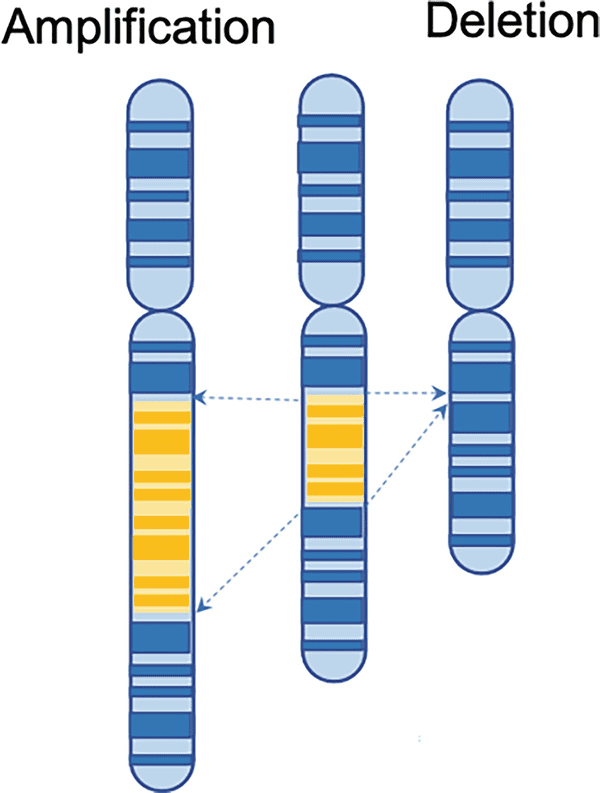
درباره CNV ها
CNV یا Copy number variation ها مجموعه ای از تغییرات ساختاری بزرگ در حد مگاباز می باشند که اخیرا نقش آن ها در بیماری های ذهنی Mental Disorders همچون اسکیزوفرنی، عقب ماندگی ذهنی و پارکینسون دیده شده است. تقریباً دو سوم کل ژنوم انسان ممکن است از تکرارها تشکیل شده باشد و 4.8 تا 9.5 درصد از ژنوم انسان را می توان به عنوان تغییرات تعداد کپی طبقه بندی کرد. تغییرات تعداد کپی (CNVs) تغییرات ژنومی هستند که منجر به تعداد غیرعادی کپی از یک یا چند ژن می شود.
مطالعات قبلی CNV را به عنوان یک قطعه DNA بزرگتر از 1000 جفت باز تعریف می کردند، اما در حال حاضر، اندازه Copy number variation ها از 50 جفت باز تا چندین مگابایت تعریف شده است. بازآرایی های ساختاری ژنومی مانند تکرارها، حذف ها، جابه جایی ها و وارونگی ها می توانند باعث ایجاد CNV شوند. CNV ها، مواد خام را برای گسترش و تنوع خانواده ژن فراهم می کند، که یک نیروی تکاملی مهم است.
علاوه بر این، واریانتهای تعداد کپی (CNVs) میتوانند بر سطوح رونویسی و ترجمه ژن نیز تأثیر بگذارند و با حساسیت به بیماریهای پیچیده مرتبط هستند. تا به امروز، تقریباً 2000 CNV توصیف شده است. ممکن است هزاران CNV دیگر در جمعیت انسانی وجود داشته باشد. جهشهای بالقوه بیماریزای DNA شامل تغییرات تک نوکلئوتیدها تا کروموزومهای کامل است. تغییرات کوچک 1 نوکلئوتیدای (nt) را SNP و تغییرات تا 50 nt در مکان واحد را تغییر کوتاه درج – حذف (indel) می نامند.
در حالی که تغییرات بزرگتر از 50 nt را واریانت های ساختاری (SVs) نامیده شده که قبلاً به عنوان تغییرات بزرگتر از 1000 nt تعریف شده است. چنین SV ها شامل درج، حذف، تکرار، وارونگی و جابجایی است. ترکیب این SVها نیز در یک ژنوم واحد امکان پذیر است. حذفها و تکرارها، که معمولاً تغییرات تعداد کپی (CNV) نامیده میشوند، به بخش بزرگی از تغییرات ژنتیکی کمک میکنند و از اهمیت تشخیصی برخوردار هستند، زیرا میتوانند نقش مهمی در ایجاد بیماریهای ژنتیکی ایفا کنند.
چندین روش مبتنی بر آزمایشگاه از جمله (MLPA) ، هیبریداسیون ژنومی مقایسهای مبتنی بر ریزآرایه (aCGH) و Arrays SNP ، توالییابی RNA هیبریداسیون درجا فلورسانس (FISH) و روش های مبتنی بر PCR برای تشخیص CNV استفاده می شود. همه این روش ها آزمایشگاهی توان عملیاتی پایینی داشته و گران هستند. در این میان، آزمایشگاههای تشخیصی معمولاً از aCGH/SNP Arrays و MLPA استفاده میکنند. روش aCGH حساس است، اما تنها محدود به تشخیص توالی های CNV موجود در ژنوم مرجع که از قبل برای طراحی پروب های آرایه استفاده شده است می باشد.
محدودیت در آزمایش های مبتنی بر MLPA تعداد پروب های موجود در کیت است که برای مالتیپلکس کردن تا حدود 50 پروب طراحی شده است، بنابراین برای یک یا چند ژن کوچکتر مناسب است. با ارائه یک نمای نوکلئوتید به نوکلئوتید از ژنوم، NGS واریانت های کوچک یا جدید CNV را که در مطالعات Array اغلب از دست می رود، تشخیص می دهد. NGS همچنین می تواند مکان دقیق یک CNV را نیز ترسیم کند. وضوح بالای توالی یابی، توان عملیاتی بالای آرایه ها را تکمیل می کند و یک دید جامع از ژنوم را به دست می دهد.
برای تشخیص CNV ها از روش های مختلفی از جمله کاریوتایپ، CGH array،FISH و غیره استفاده می شود که در این میان روش های توالی یابی نسل دوم و سوم امروزه بسیار مورد توجه قرار گرفته اند. با تکامل فناوری های نسل بعدی توالی یابی (NGS)، آزمایشگاه های تشخیص به شدت از داده های NGS در تشخیص SNP ها و ایندل ها استفاده می کنند.
علاوه بر این، NGS مزایای تشخیص دقیق موقعیت های نقطه شکست CNV در ژنوم را فراهم می کند. از این رو استفاده از NGS برای تشخیص CNV به آزمایشگاههای تشخیصی در آزمایش تعداد بیشتری از ژنها برای CNV کمک میکند. در روشهای معمول تشخیصی سنتی، نمونهها با آزمایش MLPA از ژنها بر اساس درخواستها آنالیز میشوند. از آنجایی که CNV اغلب اتفاق نمی افتد، نتایج MLPA اغلب منفی است. نتایج نشان داده شده است که استفاده از NGS در تشخیص، در مقایسه با استفاده از آزمایش مبتنی بر MLPA برای CNV ها، توان عملیاتی بهتری را با هزینه کمتر ارائه می دهد.
بنابراین MLPA عمدتاً برای تأیید نتایج حاصل از دادههای NGS که نشاندهنده CNV است استفاده میشود. استراتژی های مختلفی برای یافتن CNV ها در روش های توالی یابی از جمله WGS، WES و Targeted Sequencing وجود دارد از جمله آنالیز به وسیله روش Read pair ، آنالیز به وسیله روش Split Read آنالیز به وسیله روش Read depth و در نهایت آنالیز به وسیله روش Assembly می باشد.
در حال حاضراز چهار رویکرد مختلف نام برده برای تشخیص CNV ها در داده های NGS استفاده می شود یا به بیان دیگر تشخیص مبتنی بر نقشه برداری جفتی (PE)، تشخیص مبتنی بر خواندن تقسیم (SR)، تشخیص مبتنی بر مونتاژ جدید (DA) و تشخیص مبتنی بر عمق خواندن (RD). علاوه بر این، از رویکردهای ترکیبی نیز می توان استفاده کرد.
در میان این رویکردها، PE، SR و DA را می توان برای تشخیص انواع SV ها استفاده کرد، اما استفاده از این رویکردها نیازمند کیفیت داده بالا و سازگاری داده ها در مناطق است که اغلب کاربرد آنها را برای داده های توالی یابی کل ژنوم محدود می کند. از سوی دیگر، رویکرد RD فقط میتواند CNV ها (حذفها و تکراریها) های کوچک یا بسیار بزرگ را در همه انواع مناطق در ژنوم تشخیص دهد.
بسته به کیفیت داده، عمق پوشش، طول خواندن و مناطق ثبت شده، RD همچنین می تواند نقاط شکست دقیق را با دقت بالا تشخیص دهد. بهترین روش برای تشخیص CNV به داده های توالی یابی شده بستگی دارد. دادههای حاصل از پنل های ژنی (در این روش تنها ژن های هدف مانند اگزونهای خاص توالی یابی می شوند)، بنابراین مناطق ژنوم پیوسته نیستند.
از آنجایی که رویکرد RD از عمق خوانشی بالایی برای شناسایی CNV ها استفاده می کند، این رویکرد برای پانل های ژنی مناسب است. به دلیل deep-sequenced، دادههای پانل اغلب دارای عمق پوشش بالایی هستند، که دقت تشخیص CNV را از طریق رویکرد RD افزایش میدهد، اگرچه این واقعیت که مناطق اینترونیک در آنالیز گنجانده نشدهاند ممکن است حساسیت کمی نسبت به CNVهای خاص داده های کل ژنوم داشته باشد.