


بیماری ها
تومورهای مغزی و نخاعی در کودکان (Brain and Spinal Cord Tumors in Children) چیست؟
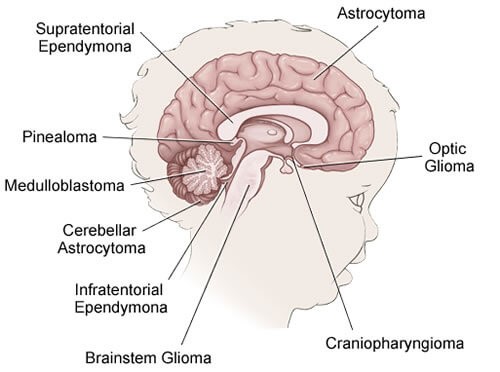
تومورهای مغزی و نخاعی در کودکان
تومورهای مغزی و نخاعی تودهای از سلولهای غیر طبیعی در مغز یا نخاع هستند که خارج از کنترل رشد کردهاند.
آیا تومورهای مغزی و نخاعی در کودکان سرطانی هستند؟
در بیشتر قسمتهای دیگر بدن، تفاوت مهمی بین تومورهای خوش خیم (غیر سرطانی) و تومورهای بدخیم (سرطانی) وجود دارد. تومورهای خوش خیم به بافتهای مجاور حمله نمیکنند و به نواحی دور گسترش نمییابند و تقریباً هرگز در سایر قسمتهای بدن تهدید کننده زندگی نیستند. تومورهای بدخیم (سرطانی) عمدتاً به این دلیل خطرناک هستند که میتوانند در سراسر بدن پخش شوند.
تومورهای مغزی به ندرت به سایر قسمتهای بدن گسترش مییابند، اگرچه بسیاری از آنها بدخیم در نظر گرفته میشوند زیرا میتوانند از طریق مغز و بافت نخاع پخش شوند. اما حتی تومورهای به اصطلاح خوش خیم میتوانند بافت طبیعی مغز را در حین رشد تحت فشار قرار دهند و از بین ببرند که همین امر میتواند منجر به آسیب جدی یا حتی گاهی تهدید کننده زندگی شود. از آن جایی که تفاوت بین تومورهای خوش خیم و بدخیم در مغز چندان مهم نیست، پزشکان معمولاً به جای «سرطان مغز» از «تومورهای مغزی» استفاده میکنند.
نگرانیهای اصلی در مورد تومورهای مغزی و نخاعی عبارتند از:
- چقدر سریع رشد میکنند.
- چقدر راحت از طریق بقیه مغز یا نخاع پخش میشوند.
- آیا میتوان آنها را برداشت یا درمان کرد و باز هم عود نکنند.
تومورهای خوش خیم و بدخیم میتوانند تهدید کننده زندگی باشند.
آیا تومورهای مغزی و نخاعی در کودکان متفاوت اند؟
تومورهای مغزی و نخاعی در کودکان با بزرگسالان متفاوت است. آنها اغلب در مکانهای مختلف تشکیل میشوند، از انواع سلولهای مختلف ایجاد میشوند و ممکن است درمان و وضعیت آتی (چشم انداز) متفاوتی داشته باشند.
سیستم عصبی مرکزی (The central nervous system)
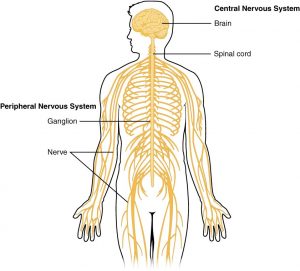
برای درک تومورهای مغزی و نخاعی، دانستن ساختار و عملکرد طبیعی سیستم عصبی مرکزی (CNS) که نام پزشکی مغز و نخاع است، به شما کمک میکند.
مغز (brain) مرکز تفکر، احساس، حافظه، گفتار، بینایی، شنوایی، حرکت و بسیاری موارد دیگر است. طناب نخاعی و اعصاب خاصی در سر به نام اعصاب جمجمه ای (cranial nerves) پیامهایی را بین مغز و بقیه بدن حمل میکنند. این پیامها به ماهیچههای ما میگویند که چگونه حرکت کنند، اطلاعات جمع آوری شده توسط حواس را منتقل کنند و به هماهنگ کردن عملکرد اندامهای داخلی کمک کنند.
مغز توسط جمجمه (skull) محافظت میشود. به همین ترتیب، نخاع (spinal cord) توسط استخوانها (مهرهها) ستون فقرات (spinal column) محافظت میشود.
مغز و نخاع توسط مایعی به نام مایع مغزی نخاعی (CSF) احاطه شده و محافظت میشوند. مایع مغزی نخاعی توسط شبکه مشیمیه (choroid plexus) ساخته میشود که در فضاهایی در مغز به نام بطن قرار دارد. بطنها و فضاهای اطراف مغز و نخاع با CSF پر میشوند.
بخشهایی از مغز و نخاع
نواحی اصلی مغز شامل مخ (cerebrum)، مخچه (cerebellum) و ساقه مغز (brain stem) است. هر قسمت عملکرد خاصی دارد.
مغ مخ: مخ بخش بزرگ و بیرونی مغز است. این قسمت به دو نیمکره یا نیمه چپ و راست (halves) تقسیم میشود و استدلال، تفکر، احساسات و زبان را کنترل میکند. همچنین مسئول حرکات ماهیچه ای برنامه ریزی شده (ارادی) (پرتاب توپ، راه رفتن، جویدن و غیره) و دریافت و تفسیر اطلاعات حسی مانند بینایی، شنوایی، بویایی، لامسه و درد است.
مخچه: مخچه در زیر مخ در قسمت پشتی مغز قرار دارد. این بخش به هماهنگی حرکت کمک میکند.
ساقه مغز: ساقه مغز قسمت پایینی مغز است که به نخاع متصل میشود. این منطقه شامل تودههایی از رشتههای عصبی بسیار طولانی است که سیگنالهایی را برای کنترل ماهیچهها و حس یا احساسات بین مغز و بقیه بدن حمل میکنند. مراکز ویژه در ساقه مغز نیز به کنترل تنفس و ضربان قلب کمک میکنند. همچنین، بیشتر اعصاب جمجمه ای (که در زیر توضیح داده شده است) از ساقه مغز شروع میشوند.
ساقه مغز به 3 قسمت اصلی تقسیم میشود: مغز میانی (midbrain)، پل مغزی (pons) و بصل النخاع (medulla oblongata).
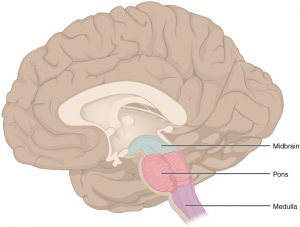
از آن جایی که ساقه مغز ناحیه کوچکی است که برای زندگی بسیار ضروری است، ممکن است نتوان با جراحی تومورهای این ناحیه را برداشت.
اعصاب جمجمه ای (Cranial nerves): اعصاب جمجمه ای مستقیماً از قاعده مغز خارج میشوند (برخلاف خروج از نخاع). این اعصاب سیگنالها را مستقیماً بین مغز و صورت، چشمها، گوشها، زبان، دهان و برخی مناطق دیگر حمل میکنند.
طناب نخاعی (Spinal cord): نخاع از دستههایی از رشتههای عصبی بسیار بلند تشکیل شده است که سیگنالهای مربوط به کنترل عضلانی، حواس یا احساسات و کنترل مثانه و روده را حمل میکنند.
انواع سلولها و بافتهای بدن در مغز و نخاع
مغز و نخاع دارای انواع مختلفی از بافتها و سلولها هستند که میتوانند به انواع مختلف تومور تبدیل شوند.
نورونها (Neurons، سلولهای عصبی): این سلولها در مغز قرار دارند که به تعیین تفکر، حافظه، احساسات، گفتار، حرکت ماهیچهها، احساس و تقریباً هر چیز دیگری که مغز و نخاع انجام میدهند، کمک میکنند.
آن ها این کار را با انتقال سیگنالهای شیمیایی و الکتریکی از طریق رشتههای عصبی خود (آکسون یا axons) انجام میدهند. آکسونهای مغز معمولا کوتاه هستند، در حالی که آکسونهای موجود در نخاع میتوانند تا چند فوت نیز طول داشته باشند.
بر خلاف بسیاری از انواع سلولهای دیگر که میتوانند رشد و تقسیم شوند تا آسیبهای ناشی از تروما یا بیماری را ترمیم کنند، سلولهای عصبی در مغز و نخاع تقریباً یک سال پس از تولد (به استثنای چند مورد) تا حد زیادی تقسیم نمیشوند. نورونها معمولاً تومور تشکیل نمیدهند اما میتوانند توسط تومورهایی که از سلولهای نزدیکشان شروع میشوند، آسیب ببینند.
سلولهای گلیال (Glial cells): سلولهای گلیال سلولهای پشتیبان مغز هستند. بیشتر تومورهای مغزی و نخاعی از سلولهای گلیال ایجاد میشوند. این تومورها گاهی اوقات به عنوان گروهی به نام گلیوما (gliomas) شناخته میشوند.
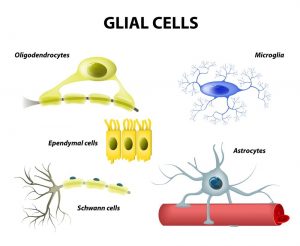
3 نوع اصلی سلولهای گلیال وجود دارد:
- آستروسیتها (Astrocytes) به حمایت و تغذیه نورونها کمک میکنند. هنگامی که مغز آسیب میبیند، آستروسیتها بافت اسکار (scar tissue) تشکیل میدهند که به ترمیم آسیب کمک میکند. تومورهای اصلی شروع شده در این سلولها آستروسیتوم (astrocytomas) یا گلیوبلاستوما (glioblastomas) نامیده میشوند.
- الیگودندروسیتها (Oligodendrocytes) میلین (myelin) را میسازند، ماده ای چرب که آکسونهای سلولهای عصبی مغز و نخاع را احاطه کرده و عایق میکند. این کار به نورونها کمک میکند سیگنالهای الکتریکی را از طریق آکسونها ارسال کنند. تومورهایی که در این سلولها شروع میشوند، الیگودندروگلیوما (oligodendrogliomas) نامیده میشوند.
- سلولهای اپاندیمال (Ependymal cells) بطنها (نواحی پر از مایع) را در قسمت مرکزی مغز میپوشانند و بخشی از مسیری را تشکیل میدهند که مایع مغزی نخاعی (CSF) از طریق آن جریان مییابد. تومورهایی که در این سلولها شروع میشوند، اپندیموم (ependymomas) نامیده میشوند.
(نوع چهارم از سلولها، به نام میکروگلیا (microglia)، سلولهای مبارزه کننده با عفونت سیستم عصبی مرکزی هستند. آنها بخشی از سیستم ایمنی هستند و سلولهای گلیال واقعی محسوب نمیشوند.)
سلولهای نورواکتودرمال (Neuroectodermal): اینها سلولها اشکال اولیه سلولهای سیستم عصبی هستند که احتمالاً در رشد سلولهای مغزی نقش دارند. آنها در سراسر مغز یافت میشوند. شایع ترین تومورهایی که از این سلولها میآیند، مدولوبلاستوما (medulloblastomas) نامیده میشوند که از مخچه منشا میگیرند.
مننژها (Meninges): لایههایی از بافت هستند که مغز و نخاع را پوشانده و از آن محافظت میکنند. مننژها به تشکیل فضاهایی کمک میکنند که CSF از طریق آنها حرکت میکند. شایع ترین تومورهایی که در این بافتها شروع میشوند، مننژیوم (meningiomas) نامیده میشوند.
شبکه کوروئید (Choroid plexus): شبکه مشیمیه ناحیه ای از مغز در داخل بطنها است که CSF را میسازد که مغز را تغذیه و محافظت میکند. تومورهایی که از این جا شروع میشوند شامل پاپیلومهای شبکه مشیمیه (choroid plexus papillomas) و کارسینومهای شبکه کوروئید (choroid plexus carcinomas) هستند.
غده هیپوفیز (Pituitary gland) و هیپوتالاموس (hypothalamus): هیپوفیز غده کوچکی در قاعده مغز است. به بخشی از مغز به نام هیپوتالاموس متصل است. هر دوی آنها هورمونهایی را میسازند که به تنظیم فعالیت چندین غده دیگر در بدن کمک میکنند. به عنوان مثال، آنها میزان هورمون تیروئید (thyroid hormone) ساخته شده توسط غده تیروئید، تولید و ترشح شیر توسط سینهها و میزان هورمونهای مردانه یا زنانه ساخته شده توسط بیضهها یا تخمدانها را کنترل میکنند. آنها همچنین هورمون رشد (growth hormone) میسازند که رشد بدن را تحریک کرده و وازوپرسین (vasopressin) که تعادل آب را توسط کلیهها تنظیم میکند.
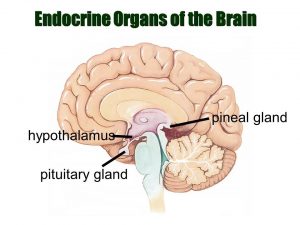
رشد تومورها در درون یا نزدیک هیپوفیز یا هیپوتالاموس و همچنین جراحی و یا پرتو درمانی در این ناحیه، میتواند بر این عملکردها تأثیر بگذارد. به عنوان مثال، تومورهایی که از غده هیپوفیز شروع میشوند، گاهی اوقات هورمون خاصی را بیش از حد تولید میکنند که میتواند مشکلاتی را ایجاد کند. از سوی دیگر، ممکن است کودک پس از روند درمان، یک یا چند هورمون را در سطح پایینی داشته باشد و برای جبران آن نیاز به مصرف هورمون داشته باشد.
غده پینه آل (Pineal gland): غده صنوبری یا پینه آل در واقع بخشی از مغز نیست. این غده یک غده درون ریز کوچک است که بین نیمکرههای مغز قرار دارد. پینه آل هورمون ملاتونین (melatonin) – هورمونی که خواب را تنظیم میکند – را در پاسخ به تغییرات نوری میسازد. شایع ترین تومورهای غده صنوبری پینئوبلاستوم (pineoblastomas) نامیده میشوند.
سد خونی-مغزی (Blood-brain barrier): پوشش داخلی رگهای خونی کوچک (مویرگها) در مغز و نخاع، یک سد بسیار انتخابی را بین خون و بافتهای سیستم عصبی مرکزی ایجاد میکند. این سد به طور معمول به حفظ تعادل متابولیک مغز کمک کرده و از ورود سموم مضر به مغز جلوگیری میکند. متأسفانه، این سد اغلب داروهای شیمی درمانی را که برای کشتن سلولهای سرطانی استفاده میشوند را از این اندامها دور نگه میدارد که در برخی موارد مفید بودن این داروها را محدود میکند.
آمار کلیدی برای تومورهای مغزی و نخاعی در کودکان
تومورهای مغزی و نخاعی دومین سرطان شایع در کودکان (پس از سرطان خون) هستند. آنها حدود 1 مورد از هر 4 سرطان دوران کودکی را تشکیل میدهند. سالانه بیش از 4000 تومور مغزی و نخاعی در کودکان و نوجوانان تشخیص داده میشود. میزان بروز (تعداد تومور در هر 100000 کودک) در سالهای اخیر تغییر چندانی نکرده است.
تومورهای بدخیم (با رشد سریع) مغز و نخاع در پسران کمی بیشتر اند در حالی که تومورهای غیر بدخیم در دختران کمی بیشتر شایع هستند.
حدود 3 کودک از هر 4 کودک مبتلا به تومور مغزی (همه انواع ترکیبی) حداقل 5 سال پس از تشخیص زنده میمانند. اما این چشم انداز ممکن است بر اساس نوع تومور، محل قرارگیری و سایر عوامل بسیار متفاوت باشد. برای دریافت اطلاعات بیشتر در مورد نرخ زندهمانی (بقا) برخی از انواع خاص تومور، به نرخ بقا برای تومورهای انتخابی مغزی و طناب نخاعی دوران کودکی مراجعه کنید.
علائم و نشانههای تومورهای مغزی و نخاعی در کودکان
علائم و نشانههای تومورهای مغزی و نخاعی ممکن است به تدریج رخ دهند و در طول زمان بدتر شوند یا حتی ممکن است به طور ناگهانی مانند تشنج رخ دهند.
علائم عمومی تومورهای مغزی و نخاعی در کودکان
وجود تومورها در هر قسمت از مغز ممکن است فشار داخل جمجمه را افزایش دهد (معروف به intracranial pressure). این اتفاق میتواند ناشی از رشد تومور، تورم در مغز یا مسدود شدن جریان مایع مغزی نخاعی باشد. افزایش فشار میتواند منجر به علائم عمومی مانند موارد زیر شود:
- سردرد
- حالت تهوع
- استفراغ
- چشمهای برگشته یا تاری دید
- مشکلات تعادلی
- تغییر رفتار
- تشنج
- خواب آلودگی یا حتی کما
سردردهایی که با گذشت زمان بدتر میشوند از علائم شایع تومورهای مغزی هستند. اما همه تومورهای مغزی باعث سردرد نمیشوند و بیشتر سردردها ناشی از وجود تومور نیستند.
در برخی از کودکان، تشنج اولین علامت تومور مغزی است. بیشتر تشنجها در کودکان ناشی از تومورهای مغزی نیستند اما اگر کودک شما تشنج داشته باشد، ممکن است پزشک کودک شما را به یک متخصص مغز و اعصاب (پزشکی که در مشکلات مغز و سیستم عصبی تخصص دارد) ارجاع دهد تا مطمئن شود که این تشنج ناشی از وجود تومور مغزی یا سایر بیماریهای جدی نیست.
در چند سال اول زندگی، سایر علائم توموری میتواند شامل موارد زیر باشد:
- تحریک پذیری
- از دست دادن اشتها
- تأخیر در رشد
- کاهش تواناییهای فکری و یا فیزیکی
- افزایش اندازه سر، گاهی همراه با ایجاد برآمدگی در نقاط نرم جمجمه (fontanelles)
در کودک مدرسه ای، سایر علائم عمومی وجود تومورها میتواند شامل عملکرد ضعیف در مدرسه، خستگی و تغییرات شخصیتی باشد.
اگر کودک بتواند همکاری کند، پزشک گاهی اوقات میتواند با نگاه کردن به داخل چشمهای کودک برای تورم عصب بینایی (معروف به papilledema) متوجه شود که آیا فشار داخل جمجمه افزایش یافته است یا خیر.
علائم وجود تومور در قسمتهای مختلف مغز یا نخاع
تومورها در قسمتهای مختلف مغز یا نخاع میتوانند علائم مختلفی را ایجاد کنند. اما این علائم میتوانند ناشی از هر گونه ناهنجاری در آن مکان خاص باشد – همیشه به این معنی نیست که کودک دارای تومور مغز یا نخاع است.
- تومورها در قسمتهایی از مغز (قسمت بزرگ و بیرونی مغز) که حرکت یا حس را کنترل میکنند، میتوانند باعث ضعف یا بیحسی در بخشی از بدن، اغلب فقط در یک طرف شوند.
- تومورها در قسمتهایی از مغز که مسئول زبان و گفتار هستند یا در نزدیکی این مناطق میتوانند در گفتار یا حتی درک کلمات مشکل ایجاد کنند.
- تومورهای قرار گرفته در قسمت جلویی مغز گاهی اوقات میتوانند بر تفکر، شخصیت و مهارتهای زبانی تأثیر بگذارند.
- تومورهای مخچه (قسمت تحتانی و پشتی مغز که هماهنگی را کنترل میکند) میتواند باعث مشکل در راه رفتن، مشکل در حرکات دقیق دستها، بازوها، پاها، مشکلات بلع یا هماهنگ سازی حرکات چشم و تغییر در ریتم گفتار شود.
- تومورها در قسمت پشتی مغز یا اطراف غده هیپوفیز، اعصاب بینایی یا برخی دیگر از اعصاب جمجمه ای میتوانند باعث بروز مشکلات بینایی شوند.
- تومورها در داخل یا نزدیک دیگر اعصاب جمجمه ای ممکن است منجر به کاهش شنوایی (در یک یا هر دو گوش)، مشکلات تعادلی، ضعف برخی از عضلات صورت، بی حسی یا درد صورت، یا مشکل در بلع شوند.
- تومورهای طناب نخاعی ممکن است باعث بی حسی، ضعف یا عدم هماهنگی در بازوها و یا پاها (معمولاً در هر دو طرف بدن) و همچنین مشکلاتی در مثانه یا روده شوند.
داشتن یک یا چند مورد از علائم بالا لزوماً به این معنی نیست که فرزند شما تومور مغزی یا نخاعی دارد. همه این علائم میتوانند دلایل دیگری نیز داشته باشند. با این حال، اگر کودک شما هر یک از این علائم را داشت، به خصوص اگر با گذشت زمان این علامت از بین نرفت یا بدتر شد، به پزشک کودک خود مراجعه کنید تا در صورت نیاز بتوان علت را پیدا کرده و آن را درمان کرد.
انواع تومورهای مغزی و نخاعی در کودکان
انواع مختلفی از تومورها میتوانند در مغز و نخاع ایجاد شوند. در حینی که پزشکان در تلاشند تا بهترین روش درمان تومور و وضعیت آتی (چشم انداز) احتمالی آن را یافته و درک کنند، چندین عامل حائز اهمیت است.
نوع تومور (بر اساس نوع سلولی که از آن شروع میشود): تومورها میتوانند تقریباً در هر نوع بافت یا سلولی در مغز یا نخاع ایجاد شوند. برخی از تومورها ترکیبی از انواع سلولها را دارند. انواع مختلف تومورها تمایل دارند تا در قسمتهای خاصی از مغز یا نخاع شروع شوند و به روشهای خاصی رشد کنند. (شایع ترین انواع تومورهای مغزی و نخاعی در کودکان در زیر توضیح داده شده است.)
درجه تومور: برخی از انواع تومورهای مغزی و نخاعی نسبت به سایرین بیشتر در بافتهای مجاور رشد میکنند (و رشد سریعی هم دارند). تومورهای مغزی و نخاعی معمولاً به 4 درجه (با استفاده از اعداد رومی I تا IV) تقسیم میشوند که عمدتاً بر اساس نحوه شکل ظاهری سلولهای تومور در زیر میکروسکوپ است. هرچه درجه بالاتر و بیشتر باشد، احتمال رشد تومور سریعتر است:
- تومورهای درجه پایین تر (درجه I یا II) تمایل به رشد آهسته تری دارند و احتمال رشد (تهاجم یا نفوذ) به بافتهای مجاور در آنها کمتر است.
- تومورهای درجه بالاتر (درجه III یا IV) تمایل به رشد سریع داشته و احتمال بیشتری برای رشد در بافتهای مجاور دارند. این تومورها اغلب نیاز به درمان شدیدتری دارند.
تغییرات ژنی موجود در سلولهای توموری: حتی برای نوع خاصی از تومور، تغییرات در ژنهای سلولهای تومور میتواند متفاوت باشد. به عنوان مثال، اکنون بسیاری از انواع تومورها بر اساس اینکه آیا سلولها دارای جهش در یکی از ژنهای IDH هستند یا خیر، تقسیم میشوند. سایر جهشهای ژنی نیز میتوانند در انواع خاصی از تومورها مهم باشند.
محل تومور: محلی که تومور در مغز و طناب نخاعی قرار دارد، میتواند روی علائمی که ایجاد میکند و همچنین درمانهایی که ممکن است برای آن مناسب باشند، تأثیر بگذارد. تومورهای مغزی در کودکان بیشتر از بزرگسالان در قسمتهای پایینی مغز مانند مخچه و ساقه مغز شروع میشوند. اما میتوانند از قسمتهای بالایی مغز نیز آغاز شوند.
گلیوما (Gliomas)
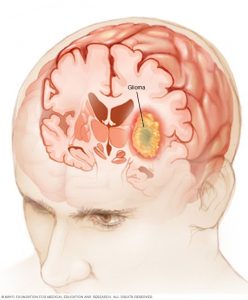
گلیوما نوع خاصی از تومور نیست. گلیوما یک اصطلاح کلی برای گروهی از تومورها است که از سلولهای گلیال (glial cells، سلولهای پشتیبان مغز) شروع میشوند. تعدادی از تومورها را میتوان گلیوما در نظر گرفت، از جمله:
- آستروسیتومها (Astrocytomas، که شامل گلیوبلاستوما میشود)
- الیگودندروگلیوما (Oligodendrogliomas)
- اپندیموما (Ependymomas)
- گلیوم ساقه مغز
- گلیوماهای بینایی
حدود نیمی از تومورهای مغزی و نخاعی در کودکان گلیوما هستند.
آستروسیتوم (Astrocytomas)
آستروسیتومها تومورهایی هستند که در سلولهایی به نام آستروسیت (astrocytes) – نوعی سلول گلیال که به حمایت و تغذیه سلولهای عصبی کمک میکند – شروع میشوند.
برخی از آستروسیتومها میتوانند به طور گسترده در سراسر مغز پخش شده و با بافت طبیعی مغز ترکیب شوند که همین امر میتواند برداشتن آنها را با جراحی سخت کند. گاهی اوقات آنها در امتداد مسیرهای مایع مغزی نخاعی (CSF) پخش میشوند. انتشار آنها به خارج از مغز یا نخاع بسیار نادر است.
مانند سایر تومورهای مغزی، آستروسیتومها اغلب بر اساس درجهشان گروه بندی میشوند.
آستروسیتومای درجه پایین (درجه I یا II) به کندی رشد میکند و شایع ترین نوع در کودکان است. برخی از انواع آن که به عنوان آستروسیتومای غیر نفوذی (non-infiltrating astrocytomas) شناخته میشوند، تومورهای درجه یکی هستند که تمایل به رشد بسیار آهستهای دارند و در بافتهای مجاور رشد نمیکنند (نفوذ نمیکنند)، بنابراین اغلب چشم انداز خوبی دارند.
- آستروسیتومای پیلوسیتیک (Pilocytic astrocytomas) تومورهای درجه یکی هستند که تمایل به رشد آهسته دارند و به ندرت در بافتهای مجاور رشد میکنند. آنها اغلب در مخچه ایجاد میشوند اما میتوانند در عصب بینایی، هیپوتالاموس، ساقه مغز یا سایر نواحی نیز شروع شوند. آنها تقریباً 1 مورد از 5 مورد تومور مغزی در کودکان را تشکیل میدهند.
- آستروسیتومای سلول غول پیکر ساب اپاندیمی (SEGAs یا Subependymal giant cell astrocytomas) در بطنها (فضاهای موجود در مغز) رخ میدهد. آنها تومورهای درجه یکی هستند که تمایل به رشد آهسته دارند و به ندرت در بافتهای مجاور رشد میکنند. این تومورها تقریباً همیشه با یک بیماری ارثی به نام توبروس اسکلروزیس (tuberous sclerosis) مرتبط هستند.
- آستروسیتومای منتشره (Diffuse astrocytomas) نیز تومورهایی با رشد آهسته هستند اما آنها تومورهای درجه II میباشند که میتوانند در بافتهای مجاور رشد کنند که همین موضوع حذف آنها را با جراحی سخت میکند. اگرچه این تومورها به عنوان تومورهای درجه پایین در نظر گرفته میشوند اما در طول زمان تهاجمی تر و سریع تر رشد میکنند.
- زانتوآستروسیتومای پلئومورفیک (PXAs یا Pleomorphic xanthoastrocytomas) تومورهای درجه II هستند که تمایل به رشد آهسته دارند و اکثر آنها تنها با جراحی قابل درمان هستند.
- گلیوماهای بینایی (Optic gliomas) آستروسیتومایی هستند که از اعصاب بینایی (اعصابی که از چشمها به مغز منتهی میشوند) شروع میشوند. آنها معمولا به کندی رشد میکنند و اغلب با یک بیماری ارثی به نام نوروفیبروماتوز (neurofibromatosis) نوع 1 مرتبط اند. این تومورها به ندرت کشنده هستند اما ممکن است باعث از دست دادن بینایی و آسیب به بافت مغز مجاور شوند.
آستروسیتومای درجه بالا (درجه III یا IV) تمایل به رشد سریع دارند و در بافت طبیعی مغز اطراف پخش میشوند. این تومورها شامل موارد زیر میشوند:
- گلیوبلاستوماها (Glioblastomas) که سریع ترین نوع آستروسیتوما (درجه IV) در حال رشد هستند.
- آستروسیتومهای آناپلاستیک (Anaplastic astrocytomas) که درجه III هستند.
الیگودندروگلیوما (Oligodendrogliomas)
این تومورها در سلولهای مغزی به نام الیگودندروسیت (نوعی سلول گلیال که مادهای چرب میسازد که به سلولهای عصبی کمک میکند تا سیگنالهای الکتریکی ارسال کنند) شروع میشوند. اینها تومورهای درجه دو هستند که تمایل به رشد آهسته دارند اما بیشتر آنها میتوانند به بافت مغز قرار گرفته در نزدیکشان رشد کنند و نمیتوان آنها را به طور کامل با جراحی خارج کرد. الیگودندروگلیوما به ندرت در امتداد مسیرهای CSF گسترش مییابد و حتی در موارد کمتری نیز در خارج از مغز یا نخاع منتشر میشود. مانند آستروسیتوما، آنها میتوانند در طول زمان تهاجمی تر شوند.
تنها حدود 1 درصد از تومورهای مغزی در کودکان الیگودندروگلیوما هستند.
اپاندیموم (Ependymomas)
حدود 5 درصد از تومورهای مغزی در کودکان اپاندیموم هستند. این تومورها از سلولهای اپاندیمی که بطنها یا کانال مرکزی نخاعی را پوشاندهاند، شروع میشوند. آنها میتوانند از تومورهای نسبتاً با درجه پایین (آهسته رشد) تا تومورهای درجه III (سریع الرشد) که اپاندیموم آناپلاستیک نامیده میشوند، متغیر باشند.
اپاندیموم ممکن است در امتداد مسیرهای CSF گسترش یابد اما به خارج از مغز یا نخاع گسترش نمییابد. این تومورها میتوانند جریان مایع مغزی نخاعی را به خارج از بطنها مسدود کنند و باعث بزرگ شدن بطنها شوند – وضعیتی به نام هیدروسفالی (hydrocephalus).
بر خلاف آستروسیتوما و اولیگودندروگلیوما، اپندیمومها معمولاً به درون بافت طبیعی مغز رشد و نفوذ نمیکنند. در نتیجه، برخی (و نه همه) اپندیمومها را میتوان با جراحی برداشت و آن را درمان کرد. اما از آن جایی که آنها میتوانند در امتداد سطوح اپاندیمی و مسیرهای CSF گسترش یابند، درمان آنها گاهی اوقات میتواند دشوار باشد.
گلیوم ساقه مغز (Brain stem gliomas)
گلیوم ساقه مغز هر نوع گلیوم است که از ساقه مغز شروع میشود. این اصطلاح به جای تومور، به محل شروع تومور اشاره دارد.
- تعداد کمی از گلیومهای ساقه مغز به صورت تومورهایی با لبههای بسیار متمایز (که گلیومهای ساقه مغز کانونی – focal brain stem gliomas – نامیده میشوند) ایجاد میشوند.
- بیشتر اوقات، گلیومهای ساقه مغز به جای رشد به عنوان یک تومور کانونی (جایی که سلولهای تومور در کنار هم قرار گرفته اند) به طور منتشر در سراسر ساقه مغز رشد میکنند (جایی که سلولهای تومور در سراسر بافت طبیعی پخش میشوند). به این تومورها گلیومهای خط میانی منتشره (diffuse midline gliomas) میگویند. این تومورها اغلب در پل مغزی شروع میشوند، جایی که به آنها گلیوماهای پونتین داخلی منتشره (DIPG یا diffuse intrinsic pontine gliomas) میگویند. درمان این تومورها ممکن است سخت باشد.
حدود 10 تا 20 درصد تومورهای مغزی در کودکان گلیوم ساقه مغز هستند. تقریباً همه این تومورها نوعی آستروسیتوم هستند.
تومورهای جنینی (Embryonal tumors)
این تومورها در اشکال اولیه سلولهای عصبی در سیستم عصبی مرکزی شروع میشوند. حدود 10 تا 20 درصد تومورهای مغزی در کودکان تومورهای جنینی هستند. آنها در کودکان کوچکتر شایع تر از بزرگترها هستند و در بزرگسالان نادر میباشند. تومورهای جنینی تمایل به رشد سریع دارند و اغلب در سراسر مسیرهای CSF گسترش مییابند.
مدولوبلاستوما (Medulloblastomas) شایع ترین نوع تومور جنینی است. این تومورها از مخچه شروع میشوند. بر اساس اینکه سلولهای تومور در زیر میکروسکوپ چگونه به نظر میرسند و سلولها دارای کدام جهشهای ژنی میباشند، انواع مختلفی از مدولوبلاستوما وجود دارد. برخی از انواع مدولوبلاستوما چشم انداز بهتری نسبت به سایرین دارند و پزشکان اکنون در تلاش هستند تا تعیین کنند چگونه این موضوع ممکن است بر درمان این تومورها تأثیر بگذارد.
مدولوبلاستوما اغلب میتواند به طور موثر درمان شود و نسبت به تومورهای جنینی در سایر قسمتهای مغز چشم انداز بهتری دارد.
سایر انواع کمتر شایع تومورهای جنینی عبارتند از:
- مدولواپیتلیوما (Medulloepithelioma)
- تومور تراتوئید (Atypical teratoid) یا رابدوئید آتیپیک ( یا rhabdoid tumor ATRT)
- تومور جنینی با روزتهای چند لایه (Embryonal tumor with multilayered rosettes)
در گذشته، بسیاری از تومورهای جنینی به عنوان تومورهای نورواکتودرمال اولیه (PNETs یا primitive neuroectodermal tumors) شناخته میشدند.
تومورهای پینه آل
برخی از انواع تومورها در غده صنوبری یا پینه آل (غده کوچکی در وسط مغز) ایجاد میشوند. شایع ترین (و سریع الرشد ترین) آنها پینئوبلاستوم (pineoblastomas) نامیده میشوند. درمان این تومورها ممکن است سخت باشد.
تومورهای سلول زاینده (Germ cell tumors) نیز که در زیر توضیح داده شده اند، میتوانند از غده صنوبری شروع شوند.
کرانیوفارنژیوم (Craniopharyngiomas)
این تومورهای با رشد آهسته در بالای غده هیپوفیز اما در زیر خود مغز شروع میشوند. آنها حدود 4 درصد از تومورهای مغزی در کودکان را تشکیل میدهند. این تومورها ممکن است به غده هیپوفیز و هیپوتالاموس فشار بیاورند و باعث ایجاد مشکلات هورمونی شوند. از آن جا که کرانیوفارنژیوم بسیار نزدیک به اعصاب بینایی شروع میشود، میتواند باعث بروز مشکلات بینایی نیز شود. این امر باعث میشود که بدون آسیب رساندن به بینایی یا تعادل هورمونی کودک، برداشتن کامل آنها سخت شود.
تومورهای گلیال و عصبی مخلوط (Mixed glial and neuronal tumors)
تومورهای خاصی که در کودکان و بزرگسالان جوان (و به ندرت در بزرگسالان مسن تر) ایجاد میشوند، دارای اجزای سلولی گلیال و عصبی هستند. آنها تمایل دارند که چشم انداز نسبتا خوبی داشته باشند.
- تومورهای عصبی اپیتلیال دیسمبریوپلاستیک (DNETs یا Dysembryoplastic neuroepithelial tumors) تمایل دارند تا تومورهایی با رشد آهسته (درجه II) باشند و بیشتر آنها تنها با جراحی قابل درمان هستند.
- گانگلیوگلیوما (Ganglioglioma) نوعی تومور درجه یک است که هم نورونهای بالغ و هم سلولهای گلیال را در بر دارد. بیشتر آنها تنها با جراحی یا با جراحی همراه با پرتو درمانی قابل درمان هستند.
تومورهای شبکه کوروئید (Choroid plexus tumors)
این تومورهای نادر از شبکه مشیمیه شروع میشوند، ناحیه ای که مایع مغزی نخاعی (CSF) را در بطنهای مغز میسازد. بیشتر آنها خوش خیم بوده (پاپیلومهای شبکه کوروئید) و با جراحی قابل درمان هستند. با این حال، برخی از آنها بدخیم میباشند (کارسینوم شبکه کوروئید یا choroid plexus carcinomas).
شوانوما (Schwannomas، نورلموما یا neurilemmomas)
این تومورها از سلولهای شوان (Schwann cells) شروع میشوند که اعصاب جمجمه و سایر اعصاب را احاطه کرده و عایق میکنند. شوانوماها معمولاً خوش خیم هستند. آنها اغلب در نزدیکی مخچه روی عصب جمجمه ای که مسئول شنوایی و تعادل است، تشکیل میشوند که در این صورت شوانوم دهلیزی (vestibular schwannomas) یا نوروم آکوستیک (acoustic neuromas) نامیده میشوند. آنها همچنین ممکن است روی اعصاب نخاعی ایجاد شوند، درست از نقطه ای که عصب از نخاع خارج میشود. در این حالت، تومور میتواند بر روی نخاع فشار بیاورد و باعث ضعف، از دست دادن حس و مشکلاتی در روده و مثانه شود.
این تومورها در کودکان نادر هستند. هنگامی که شوانوما در یک کودک یافت میشود، به خصوص اگر وی تومورهایی در دو طرف سر وجود داشته باشد، اغلب به این معنی است که کودک دارای سندرم تومور ارثی (inherited tumor syndrome) مانند نوروفیبروماتوز نوع 2 است. (به عوامل خطرزا برای تومورهای مغزی و نخاعی در کودکان مراجعه کنید.)
تومورهای دیگری که در داخل یا نزدیک مغز شروع میشوند
مننژیوم (Meningiomas)
مننژیوم تومورهای مغزی و نخاعی در کودکان
این تومورها از مننژها (meninges) شروع میشوند، لایههای بافتی که قسمت بیرونی مغز و نخاع را احاطه کرده اند. مننژیوم با فشار بر روی مغز یا نخاع علائمی را در فرد ایجاد میکند. آنها در کودکان بسیار کمتر از بزرگسالان شایع هستند.
مننژیوم تقریباً همیشه خوش خیم است و معمولاً با جراحی درمان میشود. با این حال، برخی از آنها بسیار نزدیک به ساختارهای حیاتی مغز قرار دارند و تنها با جراحی قابل درمان نیستند.
مننژیومها اغلب بر اساس ظاهر سلولهای توموری درجه بندی میشوند.
- مننژیومهای درجه یک که بیشتر شبیه سلولهای طبیعی هستند، بیشتر مننژیومها را تشکیل میدهند.
- مننژیومهای درجه دو (آتیپیک یا atypical) کمی غیر طبیعی تر به نظر میرسند.
- مننژیومهای درجه III (آناپلاستیک یا بدخیم یا malignant) که غیرطبیعی ترین سلولها به نظر میرسند، تنها حدود 1 تا 3 درصد مننژیومها را تشکیل میدهند.
احتمال عود مجدد مننژیومهای درجه بالاتر پس از درمان بیشتر است و برخی از مننژیومهای درجه III میتوانند به سایر قسمتهای بدن نیز سرایت کنند.
کوردوما (Chordomas)
این تومورها از استخوان در پایه جمجمه یا در انتهای پایین ستون فقرات شروع میشوند. کوردوماها در سیستم عصبی مرکزی شروع نمیشوند اما میتوانند با فشار آوردن بر روی قسمتهای مجاور به مغز یا نخاع آسیب برسانند. اگر این تومورها به طور کامل برداشته نشوند، تمایل به عود کردن مجدد دارند و باعث ایجاد آسیب بیشتری میشوند. این تومورها معمولاً به سایر اندامها سرایت نمیکنند. کوردوما در بزرگسالان بسیار شایع تر از کودکان است.
تومورهای سلول زایا (Germ cell tumors)
این تومورهای نادر از سلولهای زایا ایجاد میشوند که معمولاً سلولهای تخمک را در زنان و سلولهای اسپرم را در مردان تشکیل میدهند. در طول رشد طبیعی قبل از تولد، سلولهای زاینده به تخمدانها یا بیضهها میروند و به سلولهای تخمک یا اسپرم تبدیل میشوند. اما گاهی اوقات برخی از سلولهای زایا در جایی که باید حرکت نمیکنند و در مکانهای غیر طبیعی مانند مغز قرار میگیرند. سپس ممکن است به تومورهای سلول زایا تبدیل شوند، شبیه به تومورهایی که میتوانند در تخمدانها یا بیضهها تشکیل شوند.
معمولاً تومورهای سلول زایای سیستم عصبی در کودکان، اغلب در غده صنوبری یا بالای غده هیپوفیز رخ میدهند. گاهی اوقات میتوان این تومورها را بدون بیوپسی با اندازه گیری مواد شیمیایی خاص در مایع مغزی نخاعی (CSF) یا خون تشخیص داد.
انواع تومورهای سلول زاینده عبارتند از:
- ژرمینومها (Germinomas، شایع ترین نوع تومور سلول زایا CNS)
- کوریوکارسینوما (Choriocarcinomas)
- کارسینومهای جنینی (Embryonal carcinomas)
- تراتوم (Teratomas)
- تومورهای کیسه زرده (Yolk sac tumors، تومورهای سینوس اندودرمی یا endodermal sinus tumors)
نوروبلاستوم (Neuroblastomas)
این نوع تومورهای سلول عصبی سومین سرطان شایع در کودکان هستند اما نوروبلاستوم به ندرت در مغز یا نخاع ایجاد میشود. این تومورها بیشتر از سلولهای عصبی داخل شکم یا قفسه سینه ایجاد میشوند. این نوع سرطان بیشتر در اوایل دوران نوزادی دیده میشود.
لنفومها (Lymphomas)
لنفومها سرطانهایی هستند که در سلولهایی به نام لنفوسیتها (lymphocytes) شروع میشوند که گلبولهای سفید خونی هستند که بخشی از سیستم ایمنی بدن را تشکیل میدهند. بیشتر لنفومها از سایر قسمتهای بدن شروع میشوند اما بخش کوچکی از آنها در سیستم عصبی مرکزی (CNS) آغاز میشوند و به آنها لنفومهای CNS اولیه میگویند. این تومورها در کودکان نادر هستند.
تومورهای هیپوفیز
تومورهایی که از غده هیپوفیز شروع میشوند تقریباً همیشه خوش خیم (غیر سرطانی) میباشند. اما اگر آن قدر بزرگ شوند که بر ساختارهای مجاور فشار بیاورند یا هر نوع هورمونی را بیش از حد تولید کنند، باز هم میتوانند مشکلاتی را ایجاد کنند. این تومورها در نوجوانان بیشتر از کودکان کوچکتر دیده میشوند.
سرطانهایی که از سایر قسمتهای بدن به مغز سرایت میکنند
گاهی اوقات مشاهده میشود که تومورهایی از قسمت دیگری از بدن به مغز متاستاز داده اند (گسترش یافته اند). تومورهایی که از سایر اندامها شروع میشوند و سپس به مغز گسترش مییابند، تومورهای مغزی متاستاتیک (metastatic) یا ثانویه (secondary) نامیده میشوند (برخلاف تومورهای مغزی اولیه که از مغز شروع میشوند). این مسئله مهم است زیرا تومورهای متاستاتیک و اولیه مغز اغلب به طور متفاوتی درمان میشوند.
در کودکان، تومورهای مغزی متاستاتیک بسیار کمتر از تومورهای مغزی اولیه است. لوسمیهای دوران کودکی (Childhood leukemias) گاهی میتوانند به CSF اطراف مغز و نخاع سرایت کنند. هنگامی که این اتفاق میافتد، سرطان همچنان یک لوسمی در نظر گرفته میشود (سلولهای سرطانی در CSF سلولهای سرطان خون هستند)، بنابراین پزشکان از درمانهایی در زمینه سرطان خون (لوسمی) استفاده میکنند. برای اطلاعات بیشتر به لوسمی دوران کودکی مراجعه کنید.
علت ایجاد تومورهای مغزی و نخاعی در کودکان چیست؟
علت بیشتر تومورهای مغزی و نخاعی به طور کامل شناخته نشده است و عوامل خطرزا بسیار کمی برای این تومورها وجود دارد. اما محققان برخی از تغییراتی را یافته اند که در سلولهای طبیعی مغز رخ میدهد و ممکن است آنها را به تشکیل تومور سوق دهد.
سلولهای طبیعی انسان عمدتاً بر اساس اطلاعات موجود در DNA هر سلول رشد و عمل میکنند. تومورهای مغزی و نخاعی مانند سایر تومورها معمولاً در اثر تغییرات (جهش) در DNA داخل سلولها ایجاد میشوند. DNA ماده شیمیایی است که ژنهای ما را میسازد که این ژنها عملکرد سلولهای ما را کنترل میکنند. ما معمولا شبیه والدین خود هستیم زیرا آنها منبع DNA ما هستند. اما DNA بر چیزی بیشتر از ظاهر ما تأثیر میگذارد.
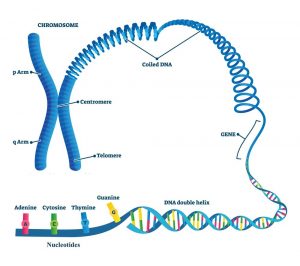
برخی از ژنها زمان رشد سلولهای ما، تقسیم به سلولهای جدید و مرگ را کنترل میکنند:
- ژنهای خاصی که به رشد، تقسیم و زنده ماندن سلولها کمک میکنند، انکوژن (oncogene) نامیده میشوند.
- ژنهایی که به کنترل تقسیم سلولی کمک میکنند یا باعث میشوند سلولها در زمان مناسب بمیرند، ژنهای سرکوب کننده تومور (tumor suppressor genes) نامیده میشوند.
سرطانها میتوانند ناشی از تغییراتی در DNA باشند که انکوژنها را فعال میکنند یا ژنهای سرکوبگر تومور را خاموش میکنند. این تغییرات ژنی میتواند از والدین به ارث برسد (همان طور که گاهی در مورد سرطانهای دوران کودکی رخ میدهد) اما اغلب در طول زندگی فرد به دست میآید.
تغییرات ژنی ارثی (Inherited gene changes)
محققان تغییرات ژنی را یافتهاند که باعث ایجاد برخی از سندرمهای ارثی نادر (مانند نوروفیبروماتوز، توبروس اسکلروزیس، سندرم Li-Fraumeni و بیماری von Hippel-Lindau) میشود و خطر ابتلا به برخی از تومورهای مغزی و نخاعی را افزایش میدهد. به عنوان مثال، سندرم Li-Fraumeni به دلیل بروز تغییرات در ژن سرکوبگر تومور TP53 ایجاد میشود. به طور معمول، این ژن از رشد سلولهای دارای DNA آسیب دیده جلوگیری میکند. تغییرات در این ژن خطر ابتلا به تومورهای مغزی (به ویژه گلیوم) و همچنین برخی سرطانهای دیگر را افزایش میدهد.
تغییرات ژنی اکتسابی (Acquired gene changes)
اغلب، مشخص نیست که چرا کودکان بدون سندرم ارثی دچار تومورهای مغزی یا نخاعی میشوند. بیشتر عوامل ایجاد کننده سرطان، مانند دود تنباکو، به نوعی به DNA آسیب میرساند. اما مغز نسبتاً در برابر بیشتر مواد شیمیایی سرطانزا که ممکن است آنها را تنفس کرده یا بخوریم، محافظت میشود. علاوه بر این، کودکان به میزان کمتری در معرض بسیاری از این مواد شیمیایی قرار میگیرند.
چندین تغییر ژنی مختلف معمولاً در سلولهای طبیعی قبل از سرطانی شدن اتفاق میافتد. انواع مختلفی از تومورهای مغزی وجود دارد که هر کدام ممکن است مجموعههای متفاوتی از تغییرات ژنی داشته باشند. تعدادی از تغییرات ژنی در انواع مختلف تومور مغزی یافت شده است اما احتمالاً بسیاری دیگر نیز وجود دارند که هنوز شناسایی نشده اند.
اکنون محققان برخی از تغییرات ژنی را که در انواع مختلف تومورهای مغزی رخ میدهد، درک میکنند اما هنوز مشخص نیست که چه چیزی باعث بروز این تغییرات میشود. برخی از تغییرات ژنی ممکن است ارثی باشد، اما اکثر تومورهای مغزی و نخاعی در کودکان نتیجه سندرمهای ارثی شناخته شده نیستند. بیشتر تغییرات ژنی احتمالاً فقط رویدادهای تصادفی هستند که گاهی اوقات در داخل سلول اتفاق میافتند، بدون اینکه علت خارجی داشته باشند.
به جز تشعشع، هیچ عامل شناخته شده مرتبط با سبک زندگی یا محیطی وجود ندارد که به وضوح با تومورهای مغزی دوران کودکی مرتبط باشد، بنابراین مهم است که به یاد داشته باشید که این کودکان یا والدین آنها هیچ کاری نمیتوانند برای پیشگیری از این سرطانها انجام دهند.
عوامل خطرزا برای تومورهای مغزی و نخاعی در کودکان
عامل خطرزا هر چیزی است که بر احتمال ابتلای فرد به بیماری مانند تومور مغزی یا نخاعی تأثیر میگذارد. انواع مختلف سرطان عوامل خطرزای متفاوتی دارند.
عوامل خطرزا مرتبط با سبک زندگی مانند رژیم غذایی، وزن بدن، فعالیت بدنی و مصرف دخانیات نقش عمده ای را در بروز بسیاری از سرطانهای بزرگسالان دارند. اما این عوامل معمولاً سالها طول میکشد تا بر خطر ابتلا به سرطان تأثیر بگذارند و تصور نمیشود که نقش زیادی در سرطانهای دوران کودکی از جمله تومورهای مغزی داشته باشند.
عوامل خطرزا بسیار کمی برای تومورهای مغزی و نخاعی یافت شده است. هیچ دلیل واضحی برای اکثر این تومورها وجود ندارد.
قرارگیری در معرض تشعشع
تنها عامل خطر محیطی ثابت شده برای تومورهای مغزی، قرار گرفتن در معرض تشعشع از ناحیه سر است که اغلب از روند درمانی سایر بیماریها ناشی میشود.
به عنوان مثال، قبل از اینکه خطرات استفاده از پرتو ها به خوبی شناخته شود (بیش از 50 سال پیش)، کودکان مبتلا به کرم حلقوی پوست سر (عفونت قارچی) اغلب پرتو درمانی با دوز پایین را دریافت میکردند. بعدها مشخص شد که این امر خطر ابتلا به برخی از انواع تومورهای مغزی را با افزایش سن افزایش میدهد.
امروزه بیشتر تومورهای مغزی ناشی از تشعشع، در اثر پرتوهایی ایجاد میشوند که به ناحیه سر برای درمان سرطانهای دیگر مانند لوسمی داده میشود. این تومورهای مغزی معمولاً حدود 10 تا 15 سال پس از پرتو درمانی ایجاد میشوند.
تومورهای ناشی از تشعشع هنوز نسبتاً نادر هستند اما به دلیل افزایش خطر (و همچنین سایر عوارض جانبی احتمالی)، پرتو درمانی تنها پس از سنجیدن دقیق مزایا و خطرات احتمالی به ناحیه سر فرد داده میشود. برای اکثر بیماران مبتلا به سرطان در داخل یا نزدیک مغز، مزایای پرتو درمانی به عنوان بخشی از درمان آنها بسیار بیشتر از خطر کوچک ایجاد تومور مغزی در سالها بعد است.
خطر احتمالی قرار گرفتن جنین یا کودک در معرض آزمایشهای تصویر برداری که از اشعه استفاده میکنند – مانند اشعه ایکس یا سی تی اسکن – به طور قطع مشخص نیست. این آزمایشها از سطوح بسیار پایینتری از تشعشعات نسبت به آنهایی که در پرتو درمانیها به کار برده میشوند، استفاده میکنند، بنابراین اگر خطری افزایش پیدا کند، احتمالاً بسیار ناچیز است. اما برای ایمن بودن، اکثر پزشکان توصیه میکنند که زنان باردار و کودکان این آزمایشها را انجام ندهند، مگر اینکه کاملاً مورد نیاز باشند.
شرایط ارثی و ژنتیکی
به ندرت، کودکان ژنهای غیر طبیعی را از والدین به ارث برده اند که آنها را در معرض افزایش خطر ابتلا به انواع خاصی از تومورهای مغزی قرار میدهد. در موارد دیگر، این ژنهای غیر طبیعی ارثی نیستند بلکه در نتیجه تغییرات (جهش) در ژن قبل از تولد رخ میدهند.
افراد مبتلا به سندرم تومور ارثی اغلب تومورهای زیادی دارند که در جوانی شروع میشوند. برخی از سندرمهای شناخته شده تر عبارتند از:
نوروفیبروماتوز (neurofibromatosis) نوع 1 (بیماری فون رکلینهاوزن یا von Recklinghausen disease)
این بیماری شایع ترین سندرم مرتبط با تومورهای مغزی یا نخاعی است. این بیماری اغلب از والدین به ارث میرسد اما میتواند در برخی از کودکانی که والدین بیماری ندارند نیز شروع شود. کودکان مبتلا به این سندرم ممکن است گلیوماهای بینایی یا سایر گلیومهای مغزی یا نخاعی یا نوروفیبروم (neurofibromas، تومورهای خوش خیم اعصاب محیطی) داشته باشند. تغییرات در ژن NF1 باعث بروز این اختلال میشود.
نوروفیبروماتوز نوع 2
این وضعیت کمتر از بیماری فون رکلینهاوزن رایج است. همچنین هم میتواند ارثی باشد و هم ممکن است در کودکان بدون سابقه خانوادگی شروع شود. این بیماری با شوانومای اعصاب جمجمه یا نخاعی، به ویژه شوانومای دهلیزی (نوروم آکوستیک) که تقریباً همیشه در هر دو طرف سر ایجاد میشود، مرتبط است. همچنین این بیماری با افزایش خطر مننژیوم و همچنین گلیوم یا اپاندیموم نخاع مرتبط است. تغییرات در ژن NF2 تقریباً همیشه مسئول بروز نوروفیبروماتوز نوع 2 است.
توبروس اسکلروزیس (Tuberous sclerosis)
کودکان مبتلا به این بیماری ممکن است به آستروسیتومای سلول غول پیکر ساب اپاندیمال (SEGAs) و همچنین سایر تومورهای خوش خیم مغز، پوست، قلب، کلیهها یا سایر اندامها مبتلا شوند. این وضعیت به دلیل تغییرات در ژن TSC1 یا TSC2 ایجاد میشود.
بیماری فون هیپل-لیندو (Von Hippel-Lindau disease)
کودکان مبتلا به این بیماری تمایل به ایجاد تومورهای عروق خونی (همانژیوبلاستوم یا hemangioblastomas) مخچه، نخاع یا شبکیه و همچنین تومورهایی در کلیه، پانکراس و برخی دیگر از قسمتهای بدن دارند. این وضعیت به دلیل تغییرات در ژن VHL ایجاد میشود.
سندرم Li-Fraumeni
افراد مبتلا به این سندرم خطر ابتلا به گلیوما و همچنین سرطان سینه، سارکوم بافت نرم (soft tissue sarcomas)، لوسمی و برخی دیگر از انواع سرطان را افزایش میدهند. این وضعیت به دلیل تغییرات در ژن TP53 ایجاد میشود.
سایر سندرمها
سایر شرایط ارثی مرتبط با افزایش خطر انواع خاصی از تومورهای مغزی و نخاعی عبارتند از:
- سندرم گورلین (Gorlin syndrome ، سندرم خال سلول بازال یا basal cell nevus syndrome)
- سندرم Turcot
- سندرم کاودن (Cowden syndrome)
- رتینوبلاستوما ارثی (Hereditary retinoblastoma)
- سندرم روبینشتاین تایبی (Rubinstein-Taybi syndrome)
برخی از خانوادهها ممکن است اختلالات ژنتیکی داشته باشند که به خوبی شناخته نشده باشد یا حتی ممکن است یک اختلال ژنتیکی مختص یک خانواده خاص باشد.
عواملی با اثرات نامشخص، بحث برانگیز یا اثبات نشده بر میزان خطر تومور مغزی
استفاده از تلفن همراه
تلفنهای همراه پرتوهای فرکانس رادیویی (RF) را منتشر میکنند که نوعی انرژی الکترومغناطیسی در طیف بین امواج رادیویی FM و امواج مورد استفاده در اجاقهای مایکروویو، رادار و ایستگاههای ماهوارهای است. تلفنهای همراه تشعشعات یونیزه تولید نمیکنند – نوعی که میتواند با آسیب رساندن به DNA داخل سلولها باعث سرطان شود. با این حال، نگرانیهایی وجود دارد که تلفنهایی که آنتنهایشان در درون آنها تعبیه شده است و بنابراین هنگام استفاده نزدیک سر قرار میگیرند، ممکن است به نوعی خطر ابتلا به تومورهای مغزی را افزایش دهند.
برخی از مطالعات احتمال افزایش خطر ابتلا به تومورهای مغزی یا شوانوم دهلیزی (vestibular schwannomas، نورومهای صوتی یا acoustic neuromas) را در بزرگسالانی که از تلفن همراه استفاده میکنند، نشان دادهاند اما بیشتر مطالعات بزرگتری که تاکنون انجام شده است، افزایش خطر را چه به طور کلی و چه در میان انواع خاصی از تومورها نشان نداده اند.
با این حال، مطالعات بسیار کمی در مورد استفاده طولانی مدت (10 سال یا بیشتر) از این دستگاهها وجود دارد و تلفنهای همراه به اندازه کافی برای تعیین خطرات احتمالی استفاده مادام العمر وجود نداشته اند. همین امر در مورد خطرات احتمالی بیشتر در کودکانی که به طور فزاینده ای از تلفن همراه استفاده میکنند، صادق است. فناوری تلفن همراه نیز همچنان در حال تغییر است و هنوز مشخص نیست که این امر چگونه ممکن است بر خطر بروز هر شرایط و وضعیتی تأثیر بگذارد.
این ریسکها در حال مطالعه هستند اما احتمالاً سالها طول خواهد کشید تا بتوان نتیجه گیری قطعی کرد. در این میان، برای افرادی که نگران خطرات احتمالی هستند، راههایی برای کاهش میزان قرار گرفتن در معرض آنها (و فرزندانشان) وجود دارد، مانند: استفاده از بلندگوی تلفن یا گوشی برای دور کردن گوشی از سر در هنگام استفاده.
عوامل دیگر
قرار گرفتن در معرض آسپارتام (جایگزین قند)، قرار گرفتن در معرض میدانهای الکترومغناطیسی از خطوط برق و سایر منابع و عفونت با ویروسهای خاص به عنوان عوامل خطرزای احتمالی پیشنهاد شده است اما اکثر محققان توافق دارند که هیچ مدرک قانع کننده ای برای ارتباط بین این عوامل با تومورهای مغزی وجود ندارد. تحقیقات در مورد این عوامل و سایر عوامل خطرزای بالقوه ادامه دارد.
آیا میتوان از بروز تومورهای مغزی و نخاعی در کودکان پیشگیری کرد؟
بزرگسالان میتوانند خطر ابتلا به برخی سرطانها را با تغییرات سبک زندگی (مانند حفظ وزن مناسب یا ترک سیگار) کاهش دهند اما در حال حاضر هیچ راه شناخته شده ای برای پیشگیری از بروز بیشتر سرطانها در کودکان وجود ندارد.
به غیر از قرار گرفتن در معرض تشعشع، هیچ عامل خطرزای مرتبط با سبک زندگی یا محیطی برای تومورهای مغزی و نخاعی در کودکان وجود ندارد، بنابراین در حال حاضر هیچ راهی برای محافظت در برابر بیشتر این سرطانها شناخته شده نیست.
محدود کردن قرار گرفتن در معرض تابش در ناحیه سر
برای اکثر کودکان مبتلا به انواع دیگر سرطان در ناحیه سر یا نزدیک سر، در صورتی که پزشکان احساس کنند فواید آن بیشتر از خطر کوچک ایجاد تومور مغزی سالها بعد باشد، ممکن است پرتو درمانی انجام شود. با این حال، در صورت نیاز، پزشکان سعی میکنند دوز پرتو را تا حد ممکن محدود کنند.
اشعه ایکس یا سی تی اسکن که قبل از تولد یا در دوران کودکی انجام میشود، نسبت به مواردی که برای درمان سرطان استفاده میشود، از اشعه بسیار کمتری استفاده میکند. اگر میزان خطرزایی این آزمایشها افزایش یابد، احتمالاً این افزایش بسیار ناچیز است اما برای ایمن بودن، اغلب پزشکان توصیه میکنند که زنان باردار و کودکان این آزمایشها را انجام ندهند، مگر اینکه کاملاً مورد نیاز باشد.
آیا تومورهای مغزی و نخاعی در کودکان به شکل زود هنگام یافت میشود؟
غربالگری، آزمایش بررسی وجود یک بیماری (مانند تومورهای مغز یا نخاع) در افراد بدون هیچ علامتی است.
در حال حاضر هیچ گونه آزمایش غربالگری به طور گسترده برای اکثر کودکان توصیه نمیشود تا به دنبال تومورهای مغزی یا نخاعی قبل از شروع علائم باشند. این تومورها معمولاً در نتیجه علائم یا علائمی که کودک به آنها دچار میشود، پیدا میشوند.
اغلب، چشم انداز کودکان مبتلا به تومورهای مغزی یا نخاعی بیشتر به نوع تومور و محل آن بستگی دارد تا اینکه چقدر زود تشخیص داده شود. اما مانند هر بیماری دیگری، تشخیص و درمان زود هنگام احتمالا مفید خواهد بود.
کودکان مبتلا به سندرمهای ارثی خاص
برای کودکان مبتلا به سندرمهای ارثی خاصی که آنها را در معرض خطر بیشتری برای ابتلا به تومورهای مغزی قرار میدهد، مانند نوروفیبروماتوز یا توبروس اسکلروزیس، پزشکان اغلب معاینات فیزیکی مکرر و آزمایشهای دیگر را توصیه میکنند.
این آزمایشات ممکن است تومورها را زمانی که هنوز کوچک هستند پیدا کنند. همه تومورهای مرتبط با این سندرمها ممکن است نیاز به درمان فوری نداشته باشند اما یافتن زود هنگام آنها ممکن است به پزشکان کمک کند تا در صورت شروع رشد یا ایجاد مشکل، به سرعت روند درمانی را آغاز کنند.
آزمایشهایی در رابطه با تومورهای مغزی و نخاعی در کودکان
تومورهای مغزی و طناب نخاعی معمولاً به دلیل علائم یا علائمی پیدا میشوند که کودک آنها را تجربه میکند. در صورت مشکوک بودن وی به داشتن تومور، آزمایشاتی برای تایید تشخیص مورد نیاز است.
تاریخچه پزشکی و معاینه فیزیکی تومورهای مغزی و نخاعی در کودکان
اگر کودک شما علائمی داشته باشد که نشان دهنده وجود تومور مغزی یا نخاعی باشد، پزشک یک تاریخچه پزشکی کامل با تمرکز بر علائم و زمان شروع آنها دریافت میکند. پزشک همچنین در صورت امکان، یک معاینه عصبی برای بررسی عملکرد مغز و نخاع کودک شما انجام میدهد. بسته به سن کودک، معاینه ممکن است رفلکس، حس، قدرت عضلانی، بینایی، حرکت چشم و دهان، هماهنگی، تعادل، هوشیاری و سایر عملکردها را آزمایش کند.
اگر نتایج غیر طبیعی باشد، پزشک کودک شما ممکن است شما را به یک متخصص مغز و اعصاب (پزشک متخصص در درمان بیماریهای سیستم عصبی) یا یک جراح مغز و اعصاب (جراح متخصص در بیماریهای سیستم عصبی) ارجاع دهد تا معاینه دقیق تری را انجام داده و حتی تستهای دیگری را تجویز کنند.
تستهای تصویر برداری
پزشکان کودک شما ممکن است یک یا چند آزمایش تصویر برداری را تجویز کنند. این آزمایشها از اشعه ایکس، آهنرباهای قوی یا مواد رادیو اکتیو برای ایجاد تصاویری از اندامهای داخلی مانند مغز و نخاع استفاده میکنند. این تصاویر ممکن است توسط پزشکان متخصص در این زمینه (جراحان مغز و اعصاب، متخصصان مغز و اعصاب و رادیولوژیستهای عصبی) و همچنین توسط پزشک مراقبتهای اولیه کودک شما بررسی شود.
تصویر برداری رزونانس مغناطیسی (Magnetic resonance imaging یا MRI) و اسکن توموگرافی کامپیوتری (computed tomography یا CT) اغلب برای بیماریهای مغزی استفاده میشوند. این اسکنها تقریباً همیشه تومور مغزی یا نخاعی را در صورت وجود نشان میدهند. پزشکان اغلب میتوانند بر اساس اینکه تومور در اسکن چگونه به نظر میرسد و در کجای مغز (یا نخاع) قرار دارد، در مورد نوع تومور نیز نظر دهند.
اسکن تصویر برداری رزونانس مغناطیسی (Magnetic resonance imaging یا MRI)
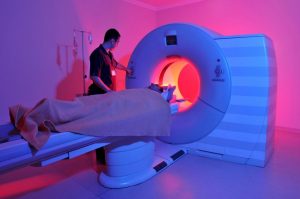
ام آر آی تومورهای مغزی و نخاعی در کودکان
اسکن MRI برای بررسی مغز و نخاع بسیار خوب است و بهترین راه برای جستجوی تومورها در این نواحی در نظر گرفته میشود. تصاویر MRI معمولاً جزئی تر از تصاویر سی تی اسکن هستند (در ادامه توضیح داده شده است). اما این روش استخوانهای جمجمه را به خوبی سی تی اسکن نشان نمیدهند و بنابراین ممکن است اثرات تومورها را بر روی جمجمه به نمایش نگذارند.
اسکنهای ام آر آی از امواج رادیویی و آهنرباهای قوی (به جای اشعه ایکس) برای ساختن تصاویر استفاده میکنند تا کودک را در معرض تشعشع قرار ندهند. ماده حاجب به نام گادولینیوم (gadolinium) ممکن است قبل از اسکن به داخل ورید تزریق شود تا به دیدن جزئیات به شکل بهتر کمک کند.
انجام اسکن ام آر آی میتواند زمان زیادی طول بکشد و لازم است فرد هر بار چند دقیقه بی حرکت بماند. برخی از کودکان ممکن است نیاز به دارو داشته باشند تا در طول آزمایش آرام شوند یا حتی بخوابند.
انواع خاصی از MRI می تواند در برخی شرایط مفید باشد:
آنژیوگرافی رزونانس مغناطیسی (Magnetic resonance angiography یا MRA) و ونوگرافی تشدید مغناطیسی (magnetic resonance venography یا MRV): این اشکال خاص MRI ممکن است برای بررسی عروق خونی مغز، به ویژه در داخل و اطراف تومور انجام شود. این کار میتواند قبل از جراحی برای کمک به جراح در جهت برنامه ریزی یک عمل بسیار مفید باشد.
طیف سنجی تشدید مغناطیسی (Magnetic resonance spectroscopy یا MRS): این آزمایش را میتوان به عنوان بخشی از MRI انجام داد. تغییرات بیوشیمیایی در ناحیه ای از مغز (که در نتایج نمودار مانند به نام طیف (spectra) نمایش داده میشود) را اندازه گیری میکند.
گاهی اوقات میتوان با مقایسه نتایج حاصل از یک تومور با بافت طبیعی مغز، به تعیین نوع تومور (یا احتمال سریع الرشد آن) کمک کرد اگرچه اغلب برای تشخیص دقیق، بیوپسی تومور هنوز مورد نیاز است. اگر آزمایش دیگری نشان داد که ناحیه ای هنوز غیر طبیعی به نظر میرسد، میتوان از MRS نیز پس از درمان استفاده کرد. MRS میتواند به تعیین اینکه آیا ناحیه تومور باقی مانده است یا احتمال بیشتری وجود دارد که بافت اسکار باشد، کمک کند.
پرفیوژن رزونانس مغناطیسی (Magnetic resonance perfusion، پرفیوژن MRI): برای این آزمایش، رنگ کنتراست (contrast dye) به سرعت به داخل ورید تزریق میشود. سپس این نوع MRI میتواند میزان خونی که از قسمتهای مختلف مغز و تومور میگذرد را نشان دهد. تومورها اغلب خون بیشتری نسبت به نواحی طبیعی مغز دارند. تومور با رشد سریعتر ممکن است به خون بیشتری نیاز داشته باشد.
ام آر آی پرفیوژن میتواند به پزشکان ایده ای را برای انتخاب بهترین مکان برای بیوپسی ارائه دهد. همچنین میتواند پس از درمان برای کمک به تعیین اینکه آیا ناحیهای که هنوز غیر طبیعی به نظر میرسد، تومور باقی مانده است یا احتمال بیشتری دارد که بافت اسکار باشد، استفاده شود.
MRI عملکردی (Functional MRI یا fMRI): این آزمایش تغییرات ریز جریان خون را در قسمت فعال مغز بررسی میکند. میتوان از آن برای تعیین اینکه کدام قسمت از مغز عملکردی مانند گفتار، فکر، احساس یا حرکت را انجام میدهد، استفاده کرد. پزشکان میتوانند از این روش برای کمک به تعیین اینکه از کدام قسمتهای مغز هنگام برنامه ریزی برای جراحی یا پرتو درمانی دوری کنند، استفاده کنند.
این تست مانند یک MRI معمولی است، با این تفاوت که از کودک شما خواسته میشود تا کارهای خاصی (مانند پاسخ دادن به سوالات ساده یا حرکت دادن انگشتان خود) را انجام دهد تا نواحی مختلف مغز را در حین انجام اسکن فعال کند.
تصویر برداری تانسور انتشار (Diffusion tensor imaging یا DTI)، همچنین به عنوان تراکتوگرافی (tractography) شناخته میشود: این روش یک نوع آزمایش MRI است که میتواند مسیرهای اصلی ماده سفید (white matter) در مغز را نشان دهد. جراحان میتوانند از این اطلاعات برای جلوگیری از برداشتن این قسمتهای مهم مغز هنگام در هنگام جراحی استفاده کنند.
اسکن توموگرافی کامپیوتری (CT یا Computed tomography)
سی تی اسکن از اشعه ایکس برای ایجاد تصاویر مقطعی دقیق از مغز و نخاع کودک شما استفاده میکند. برخلاف اشعه ایکس معمولی، سی تی اسکن تصاویر دقیقی از بافتهای نرم بدن ایجاد میکند.
برای تومورهای مغز و نخاع، سی تی اسکن به اندازه اسکن MRI استفاده نمیشود که تصاویر کمی دقیق تری ارائه میدهد و از اشعه استفاده نمیکند. با این حال، مواردی وجود دارد که سی تی اسکن ممکن است مزایایی نسبت به اسکن MRI داشته باشد:
- انجام سی تی اسکن زمان بسیار کمتری نسبت به MRI میگیرد که میتواند به ویژه برای کودکانی که در ثابت ماندن مشکل دارند، مفید باشد.
- سی تی اسکن جزئیات بیشتری از ساختارهای استخوانی نزدیک تومور را نسبت به MRI ارائه میدهد.
- سی تی آنژیوگرافی (CTA یا CT angiography) که در زیر توضیح داده شده است، میتواند جزئیات بهتری از رگهای خونی داخل و اطراف تومور نسبت به MRA در برخی موارد ارائه دهد.
قبل از اسکن، کودک شما ممکن است یک رنگ کنتراست را از طریق یک خط IV (داخل وریدی) دریافت کند. این کار به تشریح بهتر تومورهای موجود کمک میکند.
سی تی آنژیوگرافی (CTA): برای این آزمایش، کودک شما در حالی که در سی تی اسکنر قرار گرفته است، ماده حاجب را از طریق خط IV دریافت میکند. این اسکن تصاویر دقیقی را از رگهای خونی مغز ایجاد میکند که میتواند به پزشکان در روند برنامه ریزی جراحی کمک کند.
اسکن توموگرافی گسیل پوزیترون (Positron emission tomography یا PET)
برای اسکن PET، یک ماده رادیو اکتیو (معمولاً نوعی قند به نام FDG) به خون تزریق میشود. میزان رادیو اکتیویته مورد استفاده بسیار کم است و در عرض یک روز یا بیشتر از بدن خارج میشود. از آن جایی که سلولهای تومور در بدن به سرعت در حال رشد هستند، نسبت به اکثر سلولهای دیگر، مقادیر بیشتری از قند را جذب میکنند. سپس از یک دوربین مخصوص برای ایجاد تصویری از مناطق پرتوزا در بدن استفاده میشود. برخی از کودکان ممکن است نیاز به دارو داشته باشند تا در طول آزمایش آرام شوند یا حتی بخوابند.
تصویر اسکن PET به اندازه یک سی تی اسکن یا اسکن ام آر آی دقیق نیست اما میتواند اطلاعات مفیدی در مورد اینکه آیا نواحی غیر طبیعی که در آزمایشات دیگر (مانند MRI) مشاهده میشود، احتمالا تومور هستند یا خیر، ارائه دهد. این آزمایش بیشتر برای تومورهای با رشد سریع (تومورهای درجه بالا) مفید است تا برای تومورهای با رشد کندتر.
این آزمایش همچنین پس از درمان برای کمک به تعیین اینکه آیا ناحیهای که هنوز در اسکن MRI غیر طبیعی به نظر میرسد، تومور باقی مانده است یا به احتمال زیاد بافت اسکار است، مفید است. تومور باقی مانده ممکن است در اسکن PET ظاهر شود، در حالی که بافت اسکار در این روش نشان داده نمیشود.
بیوپسی (biopsy) تومور مغزی یا نخاعی
تستهای تصویر برداری مانند ام آر آی و سی تی اسکن ممکن است نشان دهند که کودک تومور مغزی یا نخاعی دارد. اما اغلب میتوان نوع تومور را تنها با برداشتن نمونه ای از آن مشخص کرد که به آن بیوپسی (biopsy) میگویند. بیوپسی ممکن است به تنهایی به عنوان یک روش برای تشخیص انجام شود یا ممکن است بخشی از جراحی برای درمان تومور باشد.
در برخی موارد (مانند بسیاری از آستروسیتومها یا گلیومهای ساقه مغز)، ممکن است بیوپسی تومور به طور ایمن ضروری یا امکان پذیر نباشد، بنابراین تشخیص تنها بر اساس ظاهر تومور در آزمایشهای تصویر برداری انجام میشود.
بیوپسی را میتوان به روشهای مختلف انجام داد.
بیوپسی سوزنی استریوتاکتیک (Stereotactic needle biopsy)
اگر آزمایشهای تصویر برداری نشان دهند که جراحی برای برداشتن تومور ممکن است بسیار خطرناک باشد (مانند برخی از تومورها در نواحی حیاتی یا اعماق مغز) ممکن است از این نوع بیوپسی استفاده شود اما برای تشخیص هنوز هم به برداشتن نمونهای نیاز است.
بسته به موقعیت، بیوپسی ممکن است در حالی که کودک بیدار یا تحت بیهوشی عمومی (خواب) باشد، انجام شود. اگر کودک بیدار باشد، جراح مغز و اعصاب یک بی حس کننده موضعی را به قسمتهایی از پوست روی جمجمه تزریق میکند تا آنها را بی حس کند. (خود جمجمه و مغز دردی را احساس نمیکنند.)
خود بیوپسی را میتوان به دو روش اصلی انجام داد:
- رایج ترین روش، گرفتن ام آر آی یا سی تی اسکن و سپس استفاده از نشانگرها (هر کدام به اندازه یک نیکل یا nickel) است که در قسمتهای مختلف پوست سر یا خطوط صورت و پوست سر برای ایجاد نقشه ای از داخل سر قرار داده شده اند. سپس یک برش (incision) در پوست سر و یک سوراخ کوچک در جمجمه ایجاد میشود. سپس از یک سیستم هدایت تصویر برای هدایت یک سوزن تو خالی به داخل تومور برای برداشتن قطعات کوچک بافت استفاده میشود.
- در رویکردی که کمتر مورد استفاده قرار میگیرد، یک قاب (frame) سفت و سخت به سر وصل میشود.
از MRI یا سی تی اسکن همراه با قاب استفاده میشود تا به جراح مغز و اعصاب کمک کند تا یک سوزن تو خالی را به داخل تومور هدایت کند تا قطعات کوچکی از بافت را بردارد. این کار هم نیاز به ایجاد یک برش در پوست سر و یک سوراخ کوچک در جمجمه دارد.
نمونههای بیوپسی سپس برای پاتولوژیست (پزشک متخصص در تشخیص بیماریها از طریق آزمایشهای آزمایشگاهی) ارسال میشوند. پاتولوژیست آن را زیر میکروسکوپ نگاه میکند (و ممکن است آزمایشهای آزمایشگاهی دیگری را انجام دهد) تا مشخص کند که آیا تومور خوش خیم یا بدخیم (سرطانی) است و دقیقاً چه نوع توموری است. این کار به تعیین بهترین دوره درمانی و وضعیت آتی (چشم انداز بیمار) کمک میکند.
کرانیوتومی (Craniotomy، بیوپسی جراحی (surgical) یا باز)
اگر آزمایشات تصویر برداری نشان دهد که تومور احتمالاً با جراحی قابل درمان است، جراح مغز و اعصاب ممکن است بیوپسی سوزنی انجام ندهد. در عوض، او ممکن است یک عمل جراحی به نام کرانیوتومی (که در جراحی تومورهای مغزی و نخاعی در کودکان توضیح داده شده است) انجام دهد تا تمام یا بیشتر تومور را خارج کند. (برداشتن بیشتر تومور به عنوان debulking شناخته میشود.)
نمونههای کوچک تومور بلافاصله توسط پاتولوژیست در حالی که کودک هنوز در اتاق عمل است، بررسی میشود تا تشخیص اولیه انجام شود. این کار میتواند به جهت دهی روند درمان کمک کند، از جمله این که آیا جراحی بیشتری باید در آن زمان انجام شود یا خیر. تشخیص نهایی در بیشتر موارد ظرف چند روز انجام میشود.
تستهای آزمایشگاهی مربوط به نمونههای بیوپسی
یافتن نوع تومور کودک برای کمک به تعیین چشم انداز (وضعیت آتی وی) و گزینههای درمانی بسیار مهم است. اما در سالهای اخیر، پزشکان دریافته اند که تغییرات در ژنها، کروموزومها یا پروتئینهای خاص در سلولهای تومور نیز میتواند مهم باشد. برخی از تومورها اکنون برای بررسی وجود این نوع تغییرات آزمایش میشوند. مثلا:
- گلیوماهایی که دارای جهشهای ژن IDH1 یا IDH2 هستند نسبت به گلیوماهای بدون این جهشهای ژنی چشم انداز بهتری دارند.
- الیگودندروگلیوماهایی که سلولهای آن بخشهایی از کروموزومهای خاصی را ندارند (که به عنوان حذف همزمان 1p19q شناخته میشود) نسبت به بیمارانی که تومورهایشان این وضعیت را ندارند، احتمالاً با شیمی درمانی درمان میشوند.
- در گلیوماهای درجه بالا، متیلاسیون پروموتر MGMT با نتایج بهتر و شانس بیشتری برای پاسخ به شیمی درمانی مرتبط است، بنابراین گاهی اوقات میتوان از آن برای کمک به روند درمانی استفاده کرد.
- برای مدولوبلاستوماها، میتوان از تغییرات در ژنهای خاص برای تقسیم این تومورها به گروههایی استفاده کرد که برخی از آنها وضعیت آتی (چشم انداز) بهتری نسبت به بقیه دارند.
.
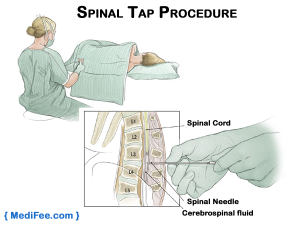
spinal tap تومورهای مغزی و نخاعی در کودکان
این آزمایش عمدتاً برای بررسی علائم سرطان در مایع مغزی نخاعی (CSF) – مایعی که مغز و نخاع را احاطه میکند – استفاده میشود. برای این آزمایش، پزشک ابتدا ناحیه ای در قسمت پایین کمر را بر روی ستون فقرات بی حس میکند. همچنین ممکن است پزشک توصیه کند که به کودک چیزی داده شود تا او را بخواباند تا این کار راحت تر و ایمن تر انجام شود. سپس یک سوزن کوچک و تو خالی بین استخوانهای ستون فقرات قرار داده میشود تا مقداری از این مایع توسط آن خارج شود.
این مایع برای بررسی وجود سلولهای سرطانی در آن زیر میکروسکوپ بررسی میشود. CSF همچنین میتواند از نظر وجود برخی مواد آزاد شده توسط برخی از تومورهای سلول زایا آزمایش شود.
اگر تومور قبلاً به عنوان نوعی که معمولاً از طریق CSF گسترش مییابد (مانند مدولوبلاستوما) تشخیص داده شده باشد، اغلب از روش سوراخهای کمری استفاده میشود. اطلاعات دریافتی از ستون فقرات میتواند بر روند درمان تأثیر بگذارد.
آسپیراسیون مغز استخوان (Bone marrow aspiration) و بیوپسی
از آن جایی که برخی از تومورها (به ویژه مدولوبلاستوماها) میتوانند فراتر از سیستم عصبی گسترش یابند، در برخی موارد پزشک ممکن است توصیه کند تا سلولهای مغز استخوان فرزندتان (قسمت نرم و داخلی برخی از استخوانها) بررسی شود تا ببینند آیا سلولهای توموری در آن جا گسترش یافته اند یا خیر.
آسپیراسیون مغز استخوان و بیوپسی اغلب همزمان انجام میشود. نمونهها معمولاً از پشت استخوان لگن (hip) گرفته میشوند اما در برخی موارد ممکن است از استخوانهای دیگری نیز گرفته شده باشند.
برای آسپیراسیون مغز استخوان، پوست روی لگن و سطح استخوان تمیز میشود و سپس با بی حسی موضعی بی حس میشود. در بیشتر موارد، به کودک داروهای دیگری نیز داده میشود تا در طول عمل، خواب آلود بوده یا حتی به خواب رود. سپس یک سوزن نازک و تو خالی به استخوان وارد میشود و از یک سرنگ برای مکیدن (اسپیراسیون) مقدار کمی از مغز استخوان مایع استفاده میشود.
بیوپسی مغز استخوان معمولاً درست بعد از آسپیراسیون انجام میشود. یک تکه کوچک از استخوان و مغز آن را با یک سوزن کمی بزرگتر که به سمت پایین و به داخل استخوان فشار داده میشود، برداشت میکنند. پس از انجام بیوپسی، برای جلوگیری از بروز خونریزی به محل فشار وارد میشود.
سپس نمونهها در زیر میکروسکوپ برای یافتن سلولهای توموری مورد بررسی قرار میگیرند.
آزمایش خون و ادرار
این آزمایشات آزمایشگاهی به ندرت برای تشخیص تومورهای مغزی و نخاعی مورد استفاده قرار میگیرند اما اگر کودک شما برای مدتی بیمار بوده است، این تستها ممکن است برای بررسی میزان عملکرد کبد، کلیهها و برخی ارگانهای دیگر انجام شوند. این امر به ویژه قبل از انجام هر گونه جراحی برنامه ریزی شده مهم است.
اگر فرزند شما تحت شیمی درمانی قرار میگیرد، آزمایش خون به طور معمول برای بررسی شمارش خون و بررسی اینکه آیا درمان بر سایر قسمتهای بدن تأثیر میگذارد یا خیر، انجام میشود.
میزان بقا (زنده مانی) برای تومورهای خاص مغز و نخاع دوران کودکی
نرخ بقا راهی برای به دست آوردن یک ایده کلی از چشم انداز (وضعیت آتی بیمار) برای افراد مبتلا به نوع خاصی از تومور است. این اطلاعات به شما میگویند چه بخشی از افراد مبتلا به همان نوع تومور تا مدت معینی (معمولاً 5 سال) پس از تشخیص هنوز زنده هستند. آنها نمیتوانند مشخص کنند که چه اتفاقی خواهد افتاد اما ممکن است به شما کمک کنند تا درک بهتری را در مورد احتمال موفقیت آمیز بودن روند درمانتان پیدا کنید.
برخی از مردم میخواهند در مورد میزان زنده مانی خود بدانند و برخی دیگر تمایلی برای دانستن این اطلاعات ندارند. اگر نمیخواهید بدانید، مجبور نیستید.
میزان بقای 5 ساله چقدر است؟
نرخ بقای 5 ساله درصد کودکانی است که حداقل 5 سال پس از تشخیص سرطان همچنان زندگی میکنند و فوت نکرده اند. به عنوان مثال، نرخ بقای 5 ساله 80 درصد به این معنی است که از هر 100 کودکی که دارای آن نوع تومور هستند، 80 نفر 5 سال پس از تشخیص هنوز زنده هستند. البته، بسیاری از کودکان بسیار بیشتر از 5 سال عمر میکنند (و حتی بسیاری از آنها درمان نیز میشوند).
نرخ بقا همه چیز را بیان نمیکند
میزان بقا اغلب بر اساس اطلاعات قبلی تعداد زیادی از افرادی است که به این بیماری مبتلا شدهاند اما پزشکان نمیتوانند پیش بینی کنند که در مورد هر کودکی چه اتفاقی خواهد افتاد. محدودیتهایی وجود دارد که باید در نظر داشت:
- اعداد زیر جزو جدیدترین اطلاعات موجود هستند. اما برای به دست آوردن نرخ بقای 5 ساله، پزشکان باید کودکانی را که حداقل 5 سال پیش تحت درمان قرار گرفته اند را بررسی کنند. همان طور که روندهای درمانی در طول زمان بهبود مییابند، کودکانی که اکنون تومورهای مغزی یا نخاعی در آنها تشخیص داده میشود ممکن است چشم انداز بهتری نسبت به این آمار داشته باشند.
- چشم انداز کودکان مبتلا به تومورهای مغزی یا نخاعی بسته به نوع تومور متفاوت است. اما بسیاری از عوامل دیگر نیز میتوانند بر وضعیت آتی کودک تأثیر بگذارند، مانند سن، محل و اندازه تومور و میزان پاسخ تومور به درمان. چشم انداز هر کودک به شرایط او بستگی دارد.
پزشک فرزند شما میتواند به شما بگوید که نرخ بقای چگونه ممکن است اعمال شود، زیرا او با وضعیت کودک شما آشنا است.
میزان بقا برای تومورهای شایع تر مغزی و نخاعی در کودکان
اعداد زیر از مرکز ثبت تومور مغزی ایالات متحده (CBTRUS) بدست آمده اند و بر اساس اطلاعات کودکان 14 ساله یا کوچکتری هستند که بین سالهای 2000 تا 2014 تحت درمان قرار گرفته اند. نکات مهمی در مورد این اعداد وجود دارد:
- این اعداد برای برخی از انواع رایج تومورها هستند. اعداد به راحتی برای همه انواع تومورهایی که در کودکان رخ میدهند در دسترس نیست، اغلب به این دلیل که نادر هستند یا طبقه بندی آنها دشوار است.
- در برخی موارد، اعداد شامل طیف وسیعی از انواع مختلف تومور است که میتوانند چشم اندازهای متفاوتی داشته باشند. به عنوان مثال، میزان بقای تومورهای شامل مدولوبلاستوماها و همچنین انواع دیگر تومورها است. مدولوبلاستوماها نسبت به سایر تومورهای جنینی چشم انداز بهتری دارند. بنابراین انتظار میرود میزان بقای واقعی مدولوبلاستوماها بیشتر از آمار موجود باشد، در حالی که تعداد سایر تومورهای جنینی احتمالاً کمتر است.
به یاد داشته باشید، این میزان بقا فقط یک آمار تخمینی است – آنها نمیتوانند پیش بینی کنند که برای هر کودکی چه اتفاقی میافتد. ما درک میکنیم که این آمار میتواند گیج کننده باشد و ممکن است شما را به سوالات بیشتری سوق دهد.
جراحی برای تومورهای مغزی و نخاعی در کودکان
برای تومورهای مغز و نخاع، جراحی ممکن است انجام شود:
- برای تعیین نوع تومور و اینکه آیا سلولهای تومور دارای تغییرات ژنی خاصی – که ممکن است بر وضعیت آتی (چشم انداز) تأثیر بگذارد – هستند، نمونه بیوپسی بگیرید.
- حذف یا تخریب تومور (یا قسمتی از آن تا حد امکان)
- کمک به پیشگیری یا درمان علائم یا عوارض احتمالی ناشی از تومور
قبل از جراحی، مطمئن شوید که هدف از جراحی چیست و همچنین فواید و خطرات احتمالی آن را درک کرده اید.
جراحی برای برداشتن یا از بین بردن تومور
اغلب، اولین گام در درمان تومور مغزی یا نخاعی این است که جراح مغز و اعصاب تا حد امکان تومور را حذف کرده یا از بین ببرد، در حالی که تلاش میکند هر گونه تأثیری را بر عملکرد طبیعی مغز یا اعصاب محدود کند.
جراحی به تنهایی یا همراه با پرتو درمانی ممکن است بسیاری از تومورهای با رشد کندتر را کنترل یا درمان کند، از جمله برخی از آستروسیتومهای با درجه پایین، تومورهای نوروپیتلیال دیسمبریوپلاستیک (DNETs یا dysembryoplastic neuroepithelial tumors)، اپاندیموما، کرانیوفارنژیوما، گانگلیوگلیوما، و مننژیوما.
کودکان مبتلا به تومورهایی که تمایل به رشد به بافت مغزی مجاور دارند، مانند آستروسیتوم آناپلاستیک یا گلیوبلاستوما، تنها با جراحی قابل درمان نیستند. اما جراحی میتواند میزان توموری را که باید با پرتو درمانی یا شیمی درمانی درمان شود، کاهش دهد که ممکن است نتایج این درمانها را بهبود بخشد.
جراحی همچنین میتواند به تسکین برخی از علائم ناشی از تومورهای مغزی، به ویژه علائم ناشی از افزایش فشار داخل جمجمه، مانند سردرد، حالت تهوع، استفراغ و تاری دید کمک کند. جراحی همچنین ممکن است کنترل تشنج را با دارو آسانتر کند.
جراحی ممکن است در برخی موارد گزینه خوبی نباشد، مانند حالتی که تومور در عمق مغز باشد یا اگر در قسمتی از مغز که قابل برداشتن نیست، مانند ساقه مغز پخش شده باشد.
اگر چنین باشد، ممکن است به جای آن از درمانهای دیگری استفاده شود.
کرانیوتومی (Craniotomy)
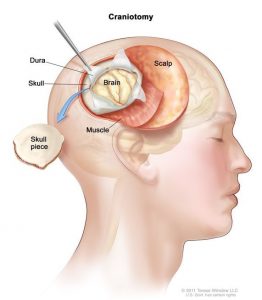
کرانیتومی تومورهای مغزی و نخاعی در کودکان
کرانیوتومی یک سوراخ جراحی است که در جمجمه ایجاد میشود. این روش رایج ترین نوع جراحی برای برداشتن تومور مغزی است. برای این عمل، اگر نیاز به ارزیابی عملکرد مغز در حین عمل باشد، ممکن است کودک یا تحت بیهوشی عمومی باشد (در خواب عمیق) یا ممکن است حداقل در بخشی از جراحی بیدار بماند ( ناحیه جراحی بی حس شده است).
ممکن است لازم باشد بخشی از سر قبل از جراحی تراشیده شود. جراح مغز و اعصاب یک برش (cut) در پوست روی جمجمه نزدیک تومور ایجاد میکند و سپس از یک مته مخصوص برای برداشتن تکه ای از استخوان از جمجمه استفاده میکند. دهانه معمولاً به اندازهای بزرگ است که جراح بتواند چندین ابزار را وارد کند و قسمتهای مورد نیاز مغز را برای یک عمل ایمن ببیند.
بسیاری از دستگاهها میتوانند به جراح کمک کنند تا تومور و بافت مغز اطراف را ببیند. جراح اغلب در حالی که مغز را از طریق میکروسکوپ نگاه میکند، عمل را انجام میدهد. تستهای تصویر برداری مانند ام آر آی یا سی تی اسکن را میتوان قبل از جراحی انجام داد (یا حتی سونوگرافی را میتوان پس از باز شدن جمجمه استفاده کرد) تا به مکان یابی تومور و لبههای آن کمک کند.
جراح تا آن جا که ممکن است تومور را از بین میبرد یا خارج میکند. بسته به سختی یا نرمی تومور و داشتن رگهای خونی زیاد یا کم، این کار را میتوان به روشهای مختلفی انجام داد:
- یکی از راهها بریدن آن با چاقوی جراحی یا قیچی مخصوص است.
- برخی از تومورها نرم هستند و با دستگاههای ساکشن قابل برداشتن میباشند.
- در موارد دیگر، یک پروب متصل به آسپیراتور اولتراسونیک (ultrasonic aspirator) را میتوان در داخل تومور قرار داد تا آن را شکسته و بمکد.
جراح بسیار مراقب است تا حد امکان از آسیب رساندن به بافت طبیعی مغز جلوگیری کند. برای کاهش خطر حذف یا آسیب به بخشهای حیاتی مغز، میتوان از تکنیکهای مختلفی استفاده کرد، مانند:
- ام آر آی عملکردی (Functional MRI): قبل از جراحی، این نوع آزمایش تصویر برداری (که در تستهای تومورهای مغزی و نخاعی در کودکان توضیح داده شده است) میتواند برای تعیین موقعیت یک عملکرد خاص از مغز انجام شود. این روش میتواند برای کمک به حفظ آن منطقه در طول عمل جراحی استفاده شود.
- تحریک قشر مغز حین عمل (نقشه برداری قشر مغز): در طول جراحی، جراح اغلب میتواند عملکرد نواحی مغز را در داخل و اطراف تومور با تحریک الکتریکی آنها و نظارت بر پاسخ تشخیص دهد. این کار نشان میدهد که آیا این مناطق عملکرد مهمی را کنترل میکنند و به جراح کمک میکند تا از آنها اجتناب کند.
- تصویر برداری حین جراحی: در برخی موارد، جراح از تصاویر MRI (یا سایر موارد) که در زمانهای مختلف در طول عمل گرفته شده است برای نشان دادن محل هر تومور باقی مانده استفاده میکند. این کار ممکن است به برخی از تومورهای مغز اجازه دهد تا با خیال راحت تر و گسترده تر برداشته شوند.
- تکنیکهای جدیدتر: انواع جدیدتر MRI و همچنین تکنیکهای دیگر مانند جراحی با هدایت فلورسانس، ممکن است در برخی شرایط مفید باشد.
پس از برداشتن تومور، جراح قطعه استخوان جمجمه را جایگزین کرده و برش را میبندد. (اگر برای بستن استخوان به پیچ، سیم یا صفحه فلزی نیاز باشد، معمولاً این وسایل از تیتانیوم ساخته شده اند که به کودک این امکان را میدهد که MRI خود را ادامه دهد و فلزیابها را خاموش نمیکند.)
برای تومورهایی که جراحی آنها سخت است، گزینه دیگر ممکن است قرار دادن یک پروب نازک با لیزر کوچک در انتهای آن از طریق سوراخ کوچکی در جمجمه و داخل تومور باشد. سپس از لیزر برای گرم کردن و از بین بردن تومور استفاده میشود. این روش که به عنوان لیزر درمانی حرارتی بینابینی (laser interstitial thermal therapy یا LITT) شناخته میشود، هنوز نسبتاً جدید است و بنابراین پزشکان هنوز در حال یادگیری بهترین راههای استفاده از آن هستند.
بعد از جراحی تومورهای مغزی و نخاعی در کودکان چه انتظاری باید داشت؟
پس از عمل برداشتن تومور، کودک ممکن است لوله ای (به نام درن یا drain) را که از برش خارج میشود و اجازه میدهد تا مایع مغزی نخاعی اضافی (CSF) از جمجمه خارج شود، داشته باشد. ممکن است لولههای دیگری در محل قرار داده شود تا خونی که پس از جراحی جمع میشود از زیر پوست سر خارج شود. زهکشها معمولاً پس از چند روز برداشته میشوند.
آزمایش تصویر برداری مانند ام آر آی یا سی تی اسکن معمولاً 1 تا 3 روز پس از عمل انجام میشود تا مشخص شود که چه مقدار از تومور برداشته شده است. زمان نقاهت در بیمارستان معمولاً 4 تا 6 روز است اما این مدت زمان به اندازه و محل تومور و اینکه آیا درمانهای دیگری انجام میشود یا خیر، بستگی دارد.
جراحی برای کمک به انسداد جریان CSF
اگر تومور جریان CSF را در سر مسدود کند، میتواند باعث افزایش فشار داخل جمجمه (ICP) شود. این حالت میتواند علائمی مانند سردرد، حالت تهوع، استفراغ و تاری دید را ایجاد کند و حتی ممکن است به مغز آسیب برساند یا تهدید کننده زندگی باشد. جراحی برای برداشتن تومور اغلب میتواند به این امر کمک کند اما راههای دیگری نیز برای تخلیه CSF اضافی و کاهش فشار در صورت نیاز وجود دارد.
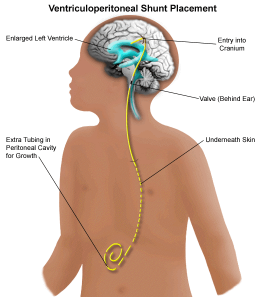
شانت تومورهای مغزی و نخاعی در کودکان
جراح مغز و اعصاب ممکن است یک لوله سیلیکونی به نام شانت (که گاهی به عنوان شانت بطنی یا VP شناخته میشود) را در مغز قرار دهد. یک انتهای شانت در یک بطن مغز (ناحیه ای پر از CSF) و سر دیگر آن در شکم یا گاهی در قلب قرار میگیرد (و سپس به عنوان شانت بطنی دهلیزی (ventriculoatrial shunt) شناخته میشود).
لوله زیر پوست گردن و قفسه سینه قرار میگیرد و اجازه میدهد CSF اضافی به داخل شکم (یا قلب) جریان یابد، جایی که با مایعات دیگر مخلوط میشود. جریان CSF توسط یک دریچه در لوله کنترل میشود.
شانتها میتوانند موقت یا دائمی باشند. آنها را میتوان قبل یا بعد از جراحی برای برداشتن تومور در بدن بیمار قرار داد. قرار دادن شانت به طور معمول حدود یک ساعت طول میکشد. اکثر کودکان باید حدود 1 تا 3 روز پس از جراحی در بیمارستان بمانند. مانند هر عمل دیگری، ممکن است عوارضی مانند خونریزی یا عفونت در فرد ایجاد شود. گاهی اوقات شانتها مسدود میشوند و نیاز به تعویض دارند.
ایجاد سوراخ در بطن سوم
گزینه دیگری برای درمان افزایش فشار در جمجمه در برخی موارد، ونتریکولوستومی سوم آندوسکوپی (endoscopic third ventriculostomy) است. در این عمل جراح در کف بطن سوم در قاعده مغز یک سوراخ ایجاد میکند تا CSF دوباره جریان یابد. این عمل از طریق یک سوراخ کوچک در جلوی جمجمه انجام میشود. مزیت این روش این است که نیازی به شانت ندارد اما این احتمال وجود دارد که سوراخ ایجاد شده در بطن دوباره بسته شود.
قرار دادن زهکش خارجی (external drain)
اگر فشار داخل سر باید برای مدت کوتاهی کاهش یابد، ممکن است یک درن بطنی خارجی (EVD یا external ventricular drain) در محل قرار داده شود تا CSF اضافی از بدن خارج شود. لوله تخلیه یک لوله کوچک است.
یک انتهای آن در یک بطن قرار میگیرد و سر دیگر به یک کیسه جمع آوری در خارج از بدن متصل میشود. همراه با جمع آوری CSF اضافی، از درن میتوان برای اندازه گیری فشار داخل جمجمه و همچنین برای جستجوی سلولهای تومور، خون یا علائم عفونت در CSF استفاده کرد.
درن را میتوان در حین جراحی یا در زمان بعد عمل در کنار تخت بیمارستان قرار داد. میتوان آن را برای کاهش فشار در روزهای قبل از جراحی یا برای کمک به تخلیه مایع جمع آوری شده پس از عمل در محل قرار داد. اگر فشار داخل جمجمه به مدت بیش از چند روز کاهش یابد، ممکن است پزشک نیاز داشته باشد که این درن را به شنت VP تغییر دهد.
جراحی برای قرار دادن کاتتر دسترسی بطنی (ventricular access catheter)
همچنین ممکن است از جراحی برای قرار دادن یک کاتتر دسترسی بطنی برای کمک به تحویل شیمی درمانی به طور مستقیم در CSF در آینده استفاده شود. یک نوع از این کاتترها مخزن اومایا (Ommaya reservoir) نامیده میشود. یک برش کوچک در پوست سر و یک سوراخ کوچک در جمجمه ایجاد میشود.
سپس یک لوله انعطاف پذیر از طریق سوراخ وارد میشود تا انتهای باز لوله در یک بطن قرار گیرد، جایی که به CSF میرسد. انتهای دیگر که دارای یک مخزن گنبدی شکل است، درست زیر پوست سر باقی میماند. پس از عمل، پزشکان و پرستاران میتوانند از یک سوزن نازک برای دادن داروهای شیمی درمانی از طریق مخزن استفاده کنند یا CSF را برای آزمایش از بطن خارج کنند.
خطرات و عوارض احتمالی جراحی تومورهای مغزی و نخاعی در کودکان
جراحی روی مغز یا نخاع یک عمل جدی است و جراحان بسیار مراقب هستند که هر گونه مشکلی را در حین یا بعد از جراحی محدود کنند. عوارض حین یا بعد از جراحی مانند خونریزی، عفونت، تشنج یا واکنش به بیهوشی نادر است اما ممکن است رخ دهد.
تورم در مغز یک نگرانی عمده پس از جراحی است. داروهایی به نام کورتیکواستروئیدها (corticosteroids) معمولاً قبل و چند روز بعد از جراحی برای کمک به کاهش این خطر تجویز میشوند.
یکی از بزرگ ترین نگرانیها هنگام برداشتن تومورهای مغزی، از دست دادن احتمالی عملکرد مغز پس از آن است، به همین دلیل است که پزشکان بسیار مراقب هستند که تنها به اندازه نیاز بافت را بردارند. هر گونه علائم آسیب مغزی پس از جراحی عمدتاً به محل و اندازه تومور بستگی دارد. اگر مشکلی پیش بیاید، ممکن است درست بعد از جراحی باشد یا حتی ممکن است روزها یا حتی هفتهها بعد باشد، بنابراین نظارت دقیق برای بررسی بروز هرگونه تغییر بسیار مهم است.
پرتو درمانی (Radiation Therapy) تومورهای مغزی و نخاعی در کودکان
پرتو درمانی از اشعه ایکس با انرژی بالا یا ذرات کوچک برای از بین بردن سلولهای سرطانی استفاده میکند. این نوع درمان توسط دکتری به نام انکولوژیست رادیو اکتیو انجام میشود.
چه زمانی میتوان از پرتو درمانی استفاده کرد؟
پرتو درمانی ممکن است در شرایط مختلف برای تومورهای مغزی یا نخاعی استفاده شود:
- اگر پس از جراحی سعی کنید سلولهای تومور باقی مانده را بکشید.
- به عنوان بخشی از درمان اصلی در صورتی که جراحی گزینه مناسبی نباشد.
- برای کمک به پیشگیری یا تسکین علائم تومور
به کودکان کمتر از 3 سال به دلیل عوارض جانبی طولانی مدت احتمالی در رشد مغز معمولاً اشعه داده نمیشود. در عوض، آنها عمدتاً با جراحی و شیمی درمانی درمان میشوند. تابش اشعه همچنین میتواند باعث ایجاد برخی مشکلات در کودکان بزرگتر نیز شود. رادیو انکولوژیستهای بسیار تلاش میکنند تا اشعه کافی را به تومور برسانند و در عین حال تابش را تا حد امکان به نواحی طبیعی اطراف مغز محدود کنند.
دریافت پرتو درمانی
بیشتر اوقات، تابش از منبعی خارج از بدن بر روی تومور متمرکز میشود. به این روش پرتو درمانی خارجی (external beam radiation therapy یا EBRT) میگویند.
قبل از شروع درمانهای کودک شما، تیم پرتو اندازی اندازه گیریهای دقیقی را برای تعیین زوایای صحیح برای نشانه گیری پرتوها و دوز مناسب تابش انجام میدهد.
این جلسه برنامه ریزی که شبیه سازی (simulation) نامیده میشود، معمولاً شامل انجام آزمایشهای تصویر برداری مانند سی تی اسکن یا اسکن MRI است. ممکن است کودک شما با یک قالب پلاستیکی مانند ثابت شود تا او را در همان موقعیت نگه دارد تا پرتو را با دقت بیشتری هدف گیری کند.
بیشتر اوقات، دوز کل تابش به بخشهای روزانه (معمولا از دوشنبه تا جمعه داده میشود) و در طول چند هفته تقسیم میشود. برای هر جلسه درمانی، کودک شما روی یک میز مخصوص دراز میکشد در حالی که یک دستگاه تابش را از زوایای دقیق ارسال میکند.
هر درمان بسیار شبیه به عکس برداری با اشعه ایکس است اما دوز پرتو بسیار بالاتر است. این روش دردناک نیست. ممکن است به برخی از کودکان کوچکتر دارو داده شود تا آنها را خواب آلود کند تا مطمئن شوند که در طول درمان حرکت نمیکنند. هر جلسه حدود 15 تا 30 دقیقه طول میکشد اما بیشتر زمان صرف اطمینان از هدف گیری صحیح پرتوها میشود. زمان واقعی درمان هر روز بسیار کوتاهتر است.
تکنیکهای خاص پرتو درمانی
پرتو درمانی میتواند به بافت طبیعی مغز آسیب برساند، بنابراین پزشکان سعی میکنند دوزهای بالایی از اشعه را با کمترین دوز ممکن به نواحی طبیعی اطراف مغز برسانند. چندین تکنیک میتواند به پزشکان کمک کند تا تابش را با دقت بیشتری متمرکز کنند:
پرتو درمانی سه بعدی منسجم (3D-CRT): 3D-CRT از نتایج آزمایشهای تصویر برداری مانند MRI و رایانههای ویژه برای نقشه برداری دقیق از محل تومور استفاده میکند. سپس چندین پرتو تشعشعی شکل میگیرد و از جهات مختلف به سمت تومور نشانه میرود. هر پرتو به تنهایی نسبتاً ضعیف است که همین امر باعث میشود کمتر به بافتهای طبیعی آسیب برساند اما پرتوها در تومور به هم میپیوندند تا دوز بالاتری از تابش را در آن جا ایجاد کنند.
پرتو درمانی تعدیل شده با شدت (IMRT): IMRT شکل پیشرفته ای از درمان سه بعدی است. علاوه بر شکل دادن به پرتوها و هدف گرفتن آنها به سمت تومور از چندین زاویه، میتوان شدت (قدرت) پرتوها را تنظیم کرد تا دوز رسیدن به حساس ترین بافتهای طبیعی را محدود کرد. این کار ممکن است به پزشک اجازه دهد دوز بالاتری را به تومور برساند. بسیاری از بیمارستانهای بزرگ و مراکز سرطان در حال حاضر از IMRT استفاده میکنند.
پرتو درمانی با پرتو پروتون منسجم (Conformal proton beam radiation therapy): درمان پرتو پروتون از رویکردی مشابه 3D-CRT استفاده میکند. اما به جای استفاده از اشعه ایکس، پرتوهای پروتون را روی تومور متمرکز میکند.
برخلاف پرتوهای ایکس که هم قبل و هم بعد از برخورد با هدف، انرژی آزاد میکنند، پروتونها آسیب کمی به بافتهایی که از آنها عبور میکنند وارد کرده و پس از طی مسافت معینی انرژی خود را آزاد میکنند. این بدان معناست که تشعشعات بیشتری را میتوان به تومور رساند و در عین حال آسیب کمتری به بافت طبیعی اطراف آن وارد کرد.
این رویکرد ممکن است برای تومورهای مغزی که لبههای مشخصی دارند (مانند کوردوما) مفیدتر باشد اما مشخص نیست که آیا برای تومورهایی که لبههای آنها با بافت طبیعی مغز مخلوط شده است (مانند آستروسیتوم یا گلیوبلاستوما) مفید باشد. در حال حاضر تعداد محدودی از مراکز پرتو پروتون در ایالات متحده وجود دارد.
رادیوسرجری استریوتاکتیک (Stereotactic radiosurgery یا SRS) یا رادیوتراپی استریوتاکتیک (SRT یا stereotactic radiotherapy): این نوع درمان دوز پرتو دهی بزرگ و دقیقی را در یک جلسه (SRS) یا در چند جلسه (SRT) به ناحیه تومور میرساند.
این روش ممکن است برای برخی از تومورها در قسمتهایی از مغز یا نخاع مفید باشد که با جراحی قابل درمان نیستند یا زمانی که کودک از سلامت کافی برای جراحی برخوردار نیست. (اصطلاح “رادیوسرجری” به این دلیل استفاده میشود که پرتو به دقت ارسال میشود اما هیچ جراحی واقعی در SRS یا SRT وجود ندارد.)
برای هر دو روش، یک قاب سر معمولاً به جمجمه متصل میشود تا به پرتوهای تشعشعی کمک کند. گاهی اوقات از ماسک صورت برای نگه داشتن سر در جای خود استفاده میشود. هنگامی که محل دقیق تومور از اسکن CT یا MRI مشخص شد، تابش از زوایای مختلف بر روی تومور متمرکز میشود. این کار به 2 روش قابل انجام است:
- در یک رویکرد، پرتوهای تابشی نازک از صدها زاویه مختلف برای مدت کوتاهی روی تومور متمرکز میشوند. هر پرتو به تنهایی ضعیف است اما همه آنها در تومور همگرا میشوند تا دوز بالاتری از تابش را ایجاد کنند. چاقوی گاما (Gamma Knife) نمونه ای از دستگاهی است که از این روش استفاده میکند.
- رویکرد دیگر از یک شتاب دهنده خطی متحرک (دستگاهی که تشعشع ایجاد میکند) استفاده میکند که توسط کامپیوتر کنترل میشود. این دستگاه به جای ارسال پرتوهای متعدد به طور همزمان، در اطراف سر حرکت میکند تا پرتو نازکی از تشعشع را از زوایای مختلف به تومور برساند. چندین دستگاه با نامهایی مانند X-Knife، CyberKnife و Clinac عمل جراحی رادیوتاکتیک استریوتاکتیک را به این روش انجام میدهند.
SRS معمولاً کل دوز تابش را در یک جلسه به فرد میتاباند، اگرچه ممکن است در صورت نیاز تکرار نیز شود.
برای SRT (که به آن رادیوسرجری تکه تکه – fractionated radiosurgery – نیز میگویند) پزشکان پرتو را در چندین دوره درمانی برای ارائه دوز مشابه یا کمی بالاتر به بیمار میدهند که اکنون اغلب بدون نیاز به قاب سر انجام میشود.
انواع دیگر پرتو درمانی
براکی تراپی (پرتو درمانی داخلی یا Brachytherapy): بر خلاف روشهای پرتو درمانی خارجی در بالا، در براکی تراپی یک منبع پرتو مستقیماً در داخل یا نزدیک تومور قرار میگیرد. تشعشعی که از آن ساطع میکند مسافت بسیار کوتاهی را طی میکند، بنابراین فقط بر تومور تأثیر میگذارد.
این تکنیک اغلب همراه با تابش خارجی استفاده میشود. این روش دوز بالایی از تشعشع را در محل تومور فراهم میکند، در حالی که تابش خارجی مناطق مجاور را با دوز کمتر تحت درمان قرار میدهد.
پرتو درمانی کل مغز و نخاع (پرتو درمانی جمجمه نخاعی یا craniospinal radiation): اگر آزمایشهایی مانند اسکن MRI یا پونکسیون کمری نشان دهد که تومور در امتداد پوشش نخاع (مننژها) یا در مایع مغزی نخاعی اطراف گسترش یافته است، ممکن است اشعه خارجی به کل مغز و نخاع داده شود. برخی از تومورها مانند اپاندیمومها و مدولوبلاستوماها به احتمال زیاد به این طریق گسترش مییابند و بنابراین ممکن است نیاز به تابش کرانیو نخاعی داشته باشند.
اثرات احتمالی پرتو درمانی
تشعشع برای سلولهای تومور مضرتر از سلولهای طبیعی است. با این حال، اشعه همچنین میتواند به بافت طبیعی مغز آسیب برساند، به ویژه در کودکان زیر 3 سال که میتواند منجر به بروز عوارض جانبی شود.
عوارض جانبی بروز یافته در طول درمان یا بلافاصله پس از درمان: در طول پرتو درمانی، برخی از کودکان ممکن است تحریک پذیر و خسته شوند. حالت تهوع، استفراغ و سردرد نیز ممکن است رخ دهد اما غیر معمول میباشد. تشعشعات ستون فقرات بیشتر از تشعشعات مغزی باعث تهوع و استفراغ میشود.
گاهی اوقات دگزامتازون (dexamethasone، یک کورتیکواستروئید) یا سایر داروها میتوانند به تسکین این علائم کمک کنند. برخی از کودکان ممکن است در مناطقی از پوست سر که پرتو دهی به سمت آنها انجام میشود، دچار ریزش مو شوند.
چند هفته پس از پرتو درمانی، کودکان ممکن است خواب آلود شوند یا سایر علائمی را در سیستم عصبی را داشته باشند. به این سندرم خواب آلودگی پرتویی (radiation somnolence syndrome) یا اثر تشعشع تاخیری زودرس (early-delayed radiation effect) میگویند. این حالت معمولا بعد از چند هفته از بین میرود.
اختلالات در تفکر و حافظه: اگر مناطق وسیعی از مغز تحت تشعشع قرار گیرد، کودکان ممکن است برخی از عملکردهای مغز خود را از دست بدهند. مشکلات میتواند شامل از دست دادن حافظه، تغییرات شخصیتی و مشکلات یادگیری در مدرسه باشد. این وضعیتها ممکن است با گذشت زمان بهتر شوند اما برخی از اثرات ممکن است طولانی مدت باشند.
سایر عوارض جانبی: سایر عوارض میتواند شامل تشنج و کند شدن رشد باشد. بسته به ناحیه ای از مغز که تحت درمان قرار گرفته و میزان اشعه داده شده ممکن است علائم دیگری نیز وجود داشته باشد.
نکروز پرتویی (Radiation necrosis): به ندرت، توده بزرگی از بافت مرده (نکروزه) در محل تومور در ماهها یا سالها پس از پرتو درمانی تشکیل میشود. اغلب میتوان آن را با داروهای کورتیکواستروئیدی کنترل کرد اما ممکن است در برخی موارد جراحی برای برداشتن بافت نکروزه مورد نیاز باشد.
افزایش خطر ایجاد تومور دیگر: تشعشع میتواند به ژنهای سلولهای طبیعی آسیب برساند. در نتیجه، خطر کمی برای ابتلا به سرطان دوم در ناحیه ای که تابش میشود وجود دارد – به عنوان مثال، مننژیوم پوششهای مغز، تومور مغزی دیگر یا به احتمال کمتر سرطان استخوان در جمجمه.
اگر این اتفاق بیفتد، معمولاً سالها از پرتو درمانی میگذرد. این خطر کوچک نباید کودکانی را که به قرار گرفتن تحت اشعه نیاز دارند از درمان باز دارد. بسیار مهم است که با پزشک فرزندتان پیگیری دقیقی را انجام دهید تا در صورت بروز مشکل، بتوان در اسرع وقت آن را پیدا و درمان کرد.
ایجاد تعادل بین خطرات و مزایا
خطر بروز تمام این عوارض جانبی باید با خطرات عدم استفاده از اشعه و کنترل کمتر تومور متعادل شود. اگر مشکلاتی پس از درمان مشاهده شود، اغلب تعیین اینکه آیا این مشکلات به دلیل آسیب ناشی از خود تومور، جراحی یا پرتو درمانی یا ترکیبی از اینها ایجاد شده است، دشوار است. پزشکان دائماً دوزهای پایینتر یا روشهای مختلف پرتو دهی را آزمایش میکنند تا ببینند آیا میتوانند به همان اندازه مؤثر باشند و مشکلات کمتری ایجاد کنند یا خیر.
سلولهای طبیعی مغز در چند سال اول زندگی به سرعت رشد میکنند و همین امر آنها را به تشعشع بسیار حساس میکند. به همین دلیل، پرتو درمانی اغلب در کودکان کمتر از 3 سال استفاده نمیشود یا به تعویق میافتد تا از بروز آسیبهایی که ممکن است بر رشد مغز تأثیر بگذارد، جلوگیری شود.
این مسئله باید با خطر رشد مجدد تومور متعادل شود زیرا پرتو درمانی اولیه ممکن است در برخی موارد نجات دهنده باشد. مهم است که با پزشک فرزندتان در مورد خطرات و مزایای درمان صحبت کنید.
شیمی درمانی (Chemotherapy) برای تومورهای مغزی و نخاعی در کودکان
شیمی درمانی (chemo) از داروهای ضد سرطانی استفاده میکند که معمولاً در ورید (IV) به فرد داده میشوند یا از طریق دهان مصرف میشوند. این داروها وارد جریان خون شده و تقریباً به تمام نواحی بدن میرسند. با این حال، بسیاری از داروهای شیمی درمانی قادر به ورود به مغز و رسیدن به سلولهای تومور نیستند.
برای برخی از تومورهای مغزی، داروها را میتوان مستقیماً در مایع مغزی نخاعی (CSF، مایعی که مغز و نخاع را احاطه میکند)، در مغز یا کانال نخاعی زیر نخاع تجویز کرد. برای کمک به این امر، یک لوله نازک به نام کاتتر دسترسی بطنی ممکن است از طریق یک سوراخ کوچک در جمجمه و در طی یک عمل جزئی وارد بطن شود. (به جراحی برای تومورهای مغزی و نخاعی در کودکان مراجعه کنید.)
چه زمانی ممکن است از شیمی درمانی استفاده شود؟
به طور کلی، شیمی درمانی برای رشد سریع تومورها استفاده میشود. برخی از انواع تومورهای مغزی، مانند مدولوبلاستوما، تمایل دارند به خوبی به شیمی درمانی پاسخ دهند.
شیمی درمانی اغلب همراه با انواع دیگر درمان مانند جراحی و پرتو درمانی استفاده میشود. این روش ممکن است به جای پرتو درمانی در کودکان 3 ساله و یا کم سن تر استفاده شود.
کدام داروهای شیمی درمانی برای درمان تومورهای مغزی و نخاعی استفاده میشود؟
برخی از داروهای شیمی درمانی مورد استفاده برای درمان کودکان مبتلا به تومورهای مغزی یا نخاعی عبارتند از:
- کربوپلاتین (Carboplatin)
- کارموستین (Carmustine یا BCNU)
- سیس پلاتین (Cisplatin)
- سیکلوفسفامید (Cyclophosphamide)
- اتوپوزید (Etoposide)
- لوموستین (Lomustine یا CCNU)
- متوترکسات (Methotrexate)
- تموزولوماید (Temozolomide)
- تیوتپا (Thiotepa)
- وین کریستین (Vincristine)
این داروها بسته به نوع تومور مغزی ممکن است به تنهایی یا در ترکیبات مختلفی با سایر داروها استفاده شوند. پزشکان شیمی درمانی را به صورت دوره ای انجام میدهند. هر چرخه معمولاً چند هفته طول میکشد و پس از آن یک دوره استراحت برای دادن زمان به بدن برای بهبودی وجود دارد.
عوارض جانبی احتمالی شیمی درمانی
داروهای شیمی درمانی میتوانند عوارض جانبی ایجاد کنند. این عوارض به نوع و دوز داروها و مدت زمان روند درمان بستگی دارد. عوارض جانبی احتمالی میتواند شامل موارد زیر باشند:
- ریزش مو
- زخمهای دهانی
- از دست دادن اشتها
- تهوع و استفراغ
- اسهال
- افزایش احتمال عفونت (به دلیل داشتن گلبولهای سفید بسیار کم)
- کبودی یا خونریزی آسان (به دلیل داشتن تعداد بسیار کم پلاکت خون)
- خستگی (به دلیل داشتن گلبولهای قرمز بسیار کم یا سایر عوامل)
برخی از مؤثرترین داروهای ضد تومورهای مغزی نسبت به سایر داروهای شیمی درمانی رایج کمتر این عوارض جانبی را دارند اما این حالات هنوز هم ممکن است رخ دهند. اکثر عوارض جانبی پس از پایان روند درمان از بین میروند. پزشک و تیم معالج فرزند شما به دقت مراقب هرگونه عارضه جانبی خواهند بود. اغلب راههایی برای کاهش این عوارض وجود دارد. به عنوان مثال، میتوان داروهایی برای کمک به پیشگیری یا کاهش تهوع و استفراغ تجویز کرد.
برخی از داروهای شیمی درمانی نیز میتوانند عوارض جانبی کمتر شایع دیگری داشته باشند. به عنوان مثال، سیس پلاتین و کربوپلاتین میتوانند باعث آسیب کلیه و کاهش شنوایی نیز شوند. اگر این داروها به کودک داده شود، پزشک کودک شما عملکرد کلیه و شنوایی او را به صورت دوره ای بررسی میکند.
حتماً از پزشک یا پرستار فرزندتان در مورد داروهایی که به کاهش عوارض جانبی کمک میکند بپرسید و به آنها اطلاع دهید که آیا کودک شما عوارض جانبی دارد تا بتوان آنها را مدیریت کرد. در برخی موارد، ممکن است لازم باشد دوز داروهای شیمی درمانی کاهش یابد یا روند درمان به تعویق بیفتد یا متوقف شود تا از بدتر شدن این اثرات جلوگیری شود.
داروهای درمانی هدفمند (Targeted Therapy Drugs) برای تومورهای مغزی و نخاعی در کودکان
همان طور که محققان در مورد تغییرات در عملکرد درونی سلولهایی که باعث سرطان میشوند یا به رشد سلولهای سرطانی کمک میکنند اطلاعات بیشتری کسب کردهاند، داروهای جدید تری تولید کردهاند که این تغییرات را هدف قرار میدهند. این داروهای هدفمند متفاوت از داروهای شیمی درمانی استاندارد عمل میکنند.
گاهی اوقات آنها زمانی که داروهای شیمی درمانی موثر نیستند، عمل میکنند و اغلب عوارض جانبی متفاوتی دارند. داروهای هدفمند هنوز نقش زیادی در درمان تومورهای مغز یا نخاع ندارند اما برخی از آنها ممکن است برای انواع خاصی از تومورها مفید باشند.
اورولیموس (Everolimus، Afinitor)
داروی هدفمند تومورهای مغزی و نخاعی در کودکان
برای آستروسیتومای سلول غول پیکر ساب اپاندیمی (SEGA) که نمیتوان آنها را به طور کامل با جراحی برداشت، اورولیموس ممکن است تومور را کوچک کرده یا رشد آن را برای مدتی کند کند. این دارو با مسدود کردن پروتئین سلولی به نام mTOR عمل میکند که به طور معمول به رشد سلولها و تقسیم به سلولهای جدید کمک میکند.
Everolimus قرصی است که یک بار در روز مصرف میشود. عوارض جانبی شایع آن عبارتند از زخمهای دهانی، افزایش خطر عفونت، حالت تهوع، از دست دادن اشتها، اسهال، زوائد پوستی، احساس خستگی یا ضعف، تجمع مایعات (معمولاً در پاها) و افزایش سطح قند و کلسترول خون. یک عارضه جانبی کمتر شایع اما جدی آسیب به ریهها است که میتواند باعث بروز تنگی نفس یا مشکلات دیگر شود.
بسیاری از داروهای هدفمند دیگر اکنون در حال توسعه و مطالعه در آزمایشات بالینی هستند.
داروهایی برای کمک به علائم در تومورهای مغزی و نخاعی در کودکان
به کودکان مبتلا به تومورهای مغزی یا نخاعی اغلب میتوان داروهایی برای کمک به کاهش علائم ناشی از وجود تومور یا عوارض جانبی ناشی از روند درمانی تجویز کرد. این داروها به طور مستقیم تومور را درمان نمیکنند اما میتوانند بخش مهمی از روند درمان کودک شما باشند.
کورتیکواستروئیدها (Corticosteroids)
داروهای شبه کورتیزون مانند دگزامتازون (Decadron) اغلب برای کاهش تورم در اطراف تومورهای مغزی تجویز میشود. آنها اغلب قبل و چند روز پس از جراحی تجویز میشوند و ممکن است در طول پرتو درمانی نیز استفاده شوند. این کار ممکن است به کاهش عوارض جانبی مانند سردرد، حالت تهوع و استفراغ کمک کند.
داروهای ضد تشنج (anticonvulsants)
ممکن است برای کاهش احتمال تشنج در کودکان مبتلا به تومور مغزی، دارو تجویز شود. بسیاری از داروهای ضد تشنج مختلف را میتوان استفاده کرد.
هورمونها
غده هیپوفیز که در پایه مغز قرار دارد به کنترل سطح بسیاری از هورمونهای مختلف در بدن کمک میکند. اگر غده هیپوفیز در اثر گسترش تومور یا روندهای درمانی (جراحی یا پرتو درمانی) آسیب دیده باشد، ممکن است کودک شما نیاز به مصرف هورمونهای هیپوفیز یا هورمونهای دیگر برای جایگزینی هورمونهایی داشته باشد که دیگر توسط بدن ساخته نمیشوند.
درمان انواع خاصی از تومورهای مغزی و نخاعی در کودکان
گزینههای درمانی برای تومورهای مغزی و نخاعی به عوامل زیادی بستگی دارد، از جمله:
- نوع تومور
- محل تومور
- این که تومور تا چه اندازه رشد کرده یا گسترش یافته است
- سن و سلامت کلی کودک
آستروسیتومای غیر نفوذی (Non-infiltrating astrocytomas، درجه I)
(آستروسیتومای پیلوسیتیک، آستروسیتومای سلول غول پیکر ساب اپاندیمی)
بسیاری از پزشکان این تومورها را تومورهای خوش خیم میدانند زیرا تمایل به رشد بسیار آهسته دارند و در بافتهای مجاور رشد نمیکنند (نفوذ میکنند). آستروسیتومای پیلوسیتیک اغلب در مخچه کودکان خردسال ایجاد میشود، در حالی که آستروسیتومای سلول غول پیکر ساب اپاندیمی (SEGAs) در بطنها رشد میکند و تقریباً همیشه در کودکان مبتلا به توبروسکلروزیس دیده میشود.
اکثر کودکان مبتلا به این آستروسیتوما تنها با جراحی قابل درمان هستند. اگر تومور به طور کامل برداشته نشود ممکن است به این تومورها پرتو درمانی داده شود، اگرچه بسیاری از پزشکان قبل از بررسی آن منتظر میمانند تا نشانههایی از رشد مجدد تومور وجود داشته باشد.
حتی در این صورت، عمل دیگری برای برداشتن تومور باقی مانده ممکن است اولین گزینه باشد. اگر تومور در مکانی باشد که اجازه نمیدهد آن را با جراحی خارج کنند – مانند هیپوتالاموس یا ساقه مغز – چشم انداز بیمار خوب نیست. در این موارد معمولا پرتو درمانی بهترین گزینه است.
برای SEGAهایی که نمیتوان آنها را به طور کامل با جراحی حذف کرد، درمان با داروی هدفمند اورولیموس (Afinitor) ممکن است تومور را کوچک کرده یا رشد آن را برای مدتی کند کند.
آستروسیتومای منتشره (Diffuse astrocytomas، درجه II)
این تومورها به آرامی رشد میکنند، اما میتوانند به بافتهای مجاور نفوذ کنند. روند درمان اولیه این تومورها در صورت وجود امکان انجام جراحی است . روند درمانی دیگر بیوپسی برای تأیید تشخیص بیماری در صورت عدم امکان انجام جراحی است. از آن جایی که این تومورها اغلب به بافت مغزی طبیعی نزدیک میشوند، درمان آنها با جراحی سخت است. معمولاً جراح سعی میکند تا جایی که ممکن است تومور را با خیال راحت خارج کند. اگر جراح بتواند همه آن را حذف کند، ممکن است کودک بدون نیاز به دوره درمانی بیشتری بهبود یابد.
پرتو درمانی ممکن است بعد از جراحی انجام شود، به خصوص اگر تومور به مقدار زیادی باقی بماند. در غیر این صورت، تابش ممکن است تا زمانی که تومور شروع به رشد مجدد کند به تعویق بیفتد. (گاهی اوقات، ممکن است قبل از دادن پرتو، یک جراحی ثانویه نیز امتحان شود.) اگر جراحی به دلیل نامناسب بودن محل تومور گزینه خوبی نباشد، ممکن است از پرتو به عنوان روند درمان اصلی استفاده شود.
برای کودکان کمتر از 3 سال، اگر تومور را نتوان به طور کامل حذف کرد یا اگر تومور دوباره رشد کند، ممکن است از شیمی درمانی برای کاهش رشد تومور تا زمانی که این کودکان بزرگ تر شوند، استفاده شود. سپس ممکن است آنها را با پرتو درمان کنند.
آستروسیتومای درجه بالاتر (درجه III یا IV)
(آستروسیتوم آناپلاستیک، گلیوبلاستوما)
اگر بتوان جراحی را انجام داد، این روش اغلب اولین روند درمان برای این آستروسیتومهای با رشد سریع است اما بیماران مبتلا به این تومورها تقریباً هرگز با جراحی درمان نمیشوند. در برخی موارد فقط بیوپسی سوزنی (needle biopsy) ایمن است. هنگامی که از جراحی استفاده میشود، تا آن جا که ممکن است تومور برداشته میشود و سپس پرتو درمانی انجام میشود که اغلب به دنبال شیمی درمانی در نظر گرفته میشود.
اگر نتوان جراحی انجام داد، پرتو درمانی درمان اصلی است که اغلب با شیمی درمانی دنبال میشود.
اگر کودک کمتر از 3 سال دارد، ممکن است تابش اشعه تا بزرگتر شدن وی به تعویق بیفتد. اگر تومور پس از دوره درمانی اولیه عود کند، ممکن است در برخی موارد جراحی تکرار شود.
از آن جایی که درمان این تومورها با روندهای درمانی فعلی سخت است، آزمایشهای بالینی دورههای درمانی نوید بخش ممکن است گزینه خوبی باشد.
الیگودندروگلیوما
در صورت امکان، جراحی اولین گزینه برای الیگودندروگلیوما است. اگرچه جراحی معمولاً این تومورهای نفوذی را درمان نمیکند اما میتواند علائم را تسکین دهد و بقا فرد را طولانی کند. بسیاری از این تومورها به کندی رشد میکنند و در صورت رشد مجدد در همان نقطه، جراحی ممکن است تکرار شود. ممکن است پس از جراحی پرتودرمانی و یا شیمی درمانی انجام شود.
اگر جراحی گزینه مناسبی نباشد، شیمی درمانی همراه با پرتو درمانی یا بدون آن، ممکن است مفید باشد. اگر سلولهای تومور تغییرات کروموزومی خاصی داشته باشند، الیگودندروگلیوما ممکن است بهتر از سایر تومورهای مغزی به شیمی درمانی پاسخ دهد. از پزشک فرزندتان در مورد وجود این تغییرات بپرسید.
اپندیموم و اپندیموم آناپلاستیک
این تومورها معمولاً به بافت مغزی طبیعی نزدیک رشد نمیکنند. اگر کل تومور برداشته شود، گاهی اوقات میتوانند با جراحی درمان شوند اما این کار همیشه امکان پذیر نیست. اگر مقداری از تومور باقی بماند، ممکن است در برخی موارد (اغلب پس از یک دوره کوتاه شیمی درمانی) عمل دوم انجام شود.
پرتو درمانی پس از جراحی برای اکثر بیماران توصیه میشود تا از عود مجدد تومور جلوگیری کنند، حتی اگر به نظر برسد که تمام تومور برداشته شده است.
استفاده از شیمی درمانی پس از جراحی هنوز در آزمایشات بالینی در حال آزمایش است. برخی از پزشکان ممکن است آن را توصیه کنند اما فایده آن هنوز نامشخص است. این کار ممکن است برای اپندیموم آناپلاستیک مفیدتر باشد. این روش ممکن است برای کمک به جلوگیری یا تأخیر در استفاده از پرتو، به کودکان بسیار خردسال پس از جراحی شیمی درمانی داده شود.
گاهی اوقات سلولهای تومور اپاندیموم میتوانند به داخل مایع مغزی نخاعی (CSF) گسترش یابند. چند هفته پس از جراحی، پزشک ممکن است اسکن MRI از مغز و نخاع را تجویز کند و CSF را برای بررسی وجود سلولهای تومور با انجام یک سوراخ کمری (تپ ستون فقرات) آزمایش کند. اگر سلولهای تومور در CSF یافت شوند یا در سطح سیستم عصبی رشد کنند، پرتو به طور معمول به کل مغز و نخاع داده میشود.
گلیوماهای بینایی
این تومورها از اعصاب بینایی (اعصابی که از چشمها به مغز منتهی میشوند) شروع میشوند.
آنها اغلب به سختی جراحی میشوند زیرا این اعصاب بسیار حساس هستند و ممکن است در اثر جراحی آسیب ببینند. بسته به محل تومور، برداشتن آن میتواند منجر به از دست دادن بینایی در یک یا هر دو چشم شود، بنابراین مزایا و خطرات جراحی باید با دقت در نظر گرفته شود. در برخی موارد، ممکن است کودک قبلاً به دلیل وجود خود تومور بینایی خود را از دست داده باشد. گاهی اوقات ممکن است نیازی به جراحی نباشد زیرا این تومورها میتوانند بسیار آهسته رشد کنند.
اگر نیاز به درمان باشد و بتوان تومور را به طور کامل برداشت، جراحی اغلب درمان ارجح است. اما در بسیاری از موارد (مخصوصاً اگر کودک مبتلا به نوروفیبروماتوز نوع 1 باشد) احتمال دارد تومور بیش از حد در امتداد اعصاب بینایی گسترش یافته باشد تا به طور کامل برداشته شود. در صورت نیاز به درمان، پرتو درمانی برای این تومورها ترجیح داده میشود، اگرچه میتواند بینایی کودک را نیز تحت تأثیر قرار دهد (و میتواند عوارض جانبی طولانی مدت دیگری نیز داشته باشد).
کودکان کوچکتر ممکن است به جای پرتو درمانی تحت شیمی درمانی قرار گیرند. در صورت نیاز، با بزرگتر شدن کودک، میتوان از پرتوها استفاده کرد.
گلیوم ساقه مغز
اکثر این تومورها آستروسیتوم هستند، اگرچه تعداد کمی از آنها اپندیموم یا سایر تومورها میباشند. این تومورها معمولاً در اسکن MRI به شکل خاصی به نظر میرسند، بنابراین تشخیص اغلب بدون جراحی یا بیوپسی انجام میشود.
گلیومهای ساقه مغز کانونی (Focal brain stem gliomas): تعداد کمی از گلیومهای ساقه مغز تومورهای کوچک با لبههای بسیار متمایز هستند (به نام گلیومهای ساقه مغز کانونی). برخی از این تومورها به قدری آهسته رشد میکنند که ممکن است نیازی به درمان نباشد مگر اینکه تومور مشکلاتی را ایجاد کند.
در صورت نیاز به درمان، این تومورها اغلب با جراحی با موفقیت درمان میشوند. اگر نتوان جراحی انجام داد، ممکن است از پرتو درمانی برای کاهش رشد آن استفاده شود. در صورتی که جراحی تومور را به طور کامل حذف نکند، میتوان از پرتو درمانی نیز استفاده کرد.
گلیومهای ساقه مغز منتشره (Diffuse brain stem gliomas): بیشتر گلیومهای ساقه مغز به جای اینکه به عنوان یک تومور متمایز (کانونی) باشند، به طور منتشره در سراسر ساقه مغز رشد میکنند. این تومورها اغلب از پونز (pons) شروع میشوند، جایی که به آنها گلیوماهای پونتین درونی منتشره (DIPG یا diffuse intrinsic pontine gliomas) میگویند.
ساقه مغز برای زندگی حیاتی است و نمیتوان آن را برداشت، بنابراین جراحی در این موارد به احتمال زیاد آسیب بیشتری را نسبت به فایده آن دارد و معمولاً انجام نمیشود. گلیومهای ساقه مغز منتشره معمولاً با پرتو درمانی درمان میشوند. گاهی اوقات شیمی درمانی به روند درمان اضافه میشود، اگرچه معلوم نیست که این کار مفید است یا خیر.
کنترل تومورهای ساقه مغزی بسیار سخت است. اما در کودکان مبتلا به نوروفیبروماتوز نوع 1، این تومورها اغلب به کندی رشد میکنند (یا حتی رشدشان متوقف میشود)، بنابراین این کودکان چشم انداز بهتری دارند. از آن جایی که درمان این تومورها سخت است، آزمایشهای بالینی روندهای درمانی نوید بخش ممکن است گزینه خوبی باشد.
تومورهای جنینی (از جمله مدولوبلاستوما)
تومورهای جنینی تمایل به رشد سریع دارند و از طریق انتشار به مایع مغزی نخاعی (CSF) گسترش مییابند. در گذشته، بسیاری از تومورهای جنینی به عنوان تومورهای نورواکتودرمال اولیه (PNETs) شناخته میشدند. تومورهای جنینی همگی به روشهای مشابهی درمان میشوند اما مدولوبلاستوماها نسبت به انواع دیگر چشم انداز بهتری دارند.
مدولوبلاستوما: این تومورها از مخچه شروع میشوند. آنها تمایل به رشد سریع دارند و از جمله مواردی هستند که به احتمال زیاد در خارج از مغز (معمولاً به استخوان یا مغز استخوان) پخش میشوند. اما آنها همچنین تمایل دارند که به خوبی به درمان پاسخ دهند.
این تومورها اغلب میتوانند جریان CSF را مسدود کنند. این اتفاق میتواند منجر به افزایش خطرناک فشار داخل جمجمه شود که ممکن است نیاز به درمان فوری با نوعی جراحی داشته باشد.
کودکان مبتلا به مدولوبلاستوما، بسته به عوامل خاصی، اغلب به دو گروه پرخطر و استاندارد تقسیم میشوند. افرادی که در گروه پرخطر قرار دارند معمولاً نسبت به کودکان گروه استاندارد درمان فشرده تری دریافت میکنند. اخیراً، پزشکان شروع به تقسیم این تومورها به 4 گروه بر اساس تغییرات ژنی در سلولهای تومور کرده اند. این تقسیم بندیها همچنین ممکن است برای کمک به تعیین بهترین درمان استفاده شوند.
مدولوبلاستوماها در صورت امکان با عمل جراحی برداشته میشوند و به دنبال آن پرتو درمانی در ناحیه ای که شروع شده است، انجام میشود. دوزهای بالای تابش به ناحیه تومور هدایت میشود. از آن جایی که این تومورها تمایل به گسترش به CSF دارند، به کودکان 3 ساله یا بزرگتر نیز ممکن است دوزهای کمتری از تابش به کل مغز و نخاع داده شود (تابش کرانیو نخاعی).
شیمی درمانی معمولاً پس از پرتو درمانی انجام میشود که ممکن است در برخی موارد به پزشکان اجازه دهد تا از دوزهای کمتری از اشعه استفاده کنند. اما اگر تومور از طریق CSF گسترش یافته باشد، دوز استاندارد پرتو مورد نیاز خواهد بود.
پزشکان سعی میکنند تا حد امکان از اشعه کمتری برای کودکان کمتر از 3 سال، استفاده کنند. شیمی درمانی معمولاً اولین دوره درمانی است که پس از جراحی انجام میشود. بسته به نحوه پاسخ تومور، شیمی درمانی ممکن است با پرتو درمانی دنبال شود.
برخی گزارشها وجود دارد که انجام شیمی درمانی با دوز بالا و به دنبال آن پیوند سلولهای بنیادی اتولوگ (autologous stem cell transplant) ممکن است برای برخی از کودکان مبتلا به مدولوبلاستوما مفید باشد. اکنون چندین آزمایش بالینی در حال مطالعه این موضوع هستند.
سایر تومورهای جنینی و پینئوبلاستوم: انواع کمتر شایع تومورهای جنینی عبارتند از:
- مدولواپیتلیوما (Medulloepithelioma)
- تومور تراتوئید/رابدوئید آتیپیک (ATRT)
- تومور جنینی با روزتهای چند لایه
Pineoblastomas دیگر نوعی تومور جنینی محسوب نمیشود اما به روشی مشابه درمان میشود.
این تومورها همچنین تمایل به رشد سریع دارند و معمولاً درمان آنها سخت تر از مدولوبلاستوما است (اگرچه درمان اغلب مانند درمان مدولوبلاستوماهای پرخطر است).
جراحی روش درمان اصلی این تومورها است اما معمولاً به سختی میتوان این تومورها را به طور کامل حذف کرد. با این حال، جراحی میتواند علائم را تسکین دهد و ممکن است به موثرتر کردن سایر دورههای درمانی نیز کمک کند. کودکان 3 ساله یا بزرگتر پس از جراحی تحت پرتو درمانی قرار میگیرند. از آن جایی که این تومورها تمایل به گسترش به CSF دارند، پرتو درمانی اغلب به کل مغز و نخاع داده میشود (اشعه کرانیو نخاعی).
شیمی درمانی ممکن است همراه با پرتو درمانی انجام شود تا بتوان از دوز کمتری از اشعه استفاده کرد. اما اگر تومور به CSF گسترش یافته باشد، دوز استاندارد تابش مورد نیاز خواهد بود. شیمی درمانی همچنین برای درمان تومورهایی که عود میکنند (recur) استفاده میشود.
پزشکان سعی میکنند تا حد امکان از اشعه کمتری برای کودکان کمتر از 3 سال، استفاده کنند. شیمی درمانی معمولاً اولین درمانی است که پس از جراحی انجام میشود. برخی از مطالعات نتایج بسیار خوبی را با استفاده از شیمی درمانی در کودکان خردسال نشان داده اند. بسته به نحوه پاسخ تومور، شیمی درمانی ممکن است با پرتو درمانی همراه باشد یا نباشد.
برخی گزارشها وجود دارد که انجام شیمی درمانی با دوز بالا به دنبال پیوند سلولهای بنیادی اتولوگ ممکن است برای کودکان مبتلا به پینئوبلاستوم و سایر انواع تومورهای جنینی مفید باشد. اکنون چندین آزمایش بالینی در حال مطالعه این موضوع هستند.
مننژیوم
جراحی روش درمان اصلی این تومورها است. اگر این روش تومور را به طور کامل از بین ببرد، معمولاً کودکان بهبود مییابند.
برخی از تومورها، به ویژه آنهایی که در پایه مغز قرار دارند، نمیتوانند به طور کامل برداشته شوند. همچنین برخی تهاجمی هستند و حتی اگر تصور میشد به طور کامل برداشته شوند، باز هم باز میگردند. پرتو درمانی پس از جراحی ممکن است رشد این تومورها را کنترل کند. اگر جراحی و پرتو درمانی مؤثر نباشند ممکن است شیمی درمانی انجام شود اما این روش در بسیاری از موارد مفید نیست.
شوانوما (از جمله نورومای آکوستیک)
این تومورهای با رشد آهسته معمولاً خوش خیم هستند و با جراحی درمان میشوند. در برخی از مراکز، شوانومای دهلیزی کوچک (همچنین به عنوان نوروم آکوستیک شناخته میشود) با رادیوسرجری استریوتاکتیک درمان میشوند. برای شوانومای بزرگتر که برداشتن کامل آنها احتمالاً مشکلاتی را ایجاد میکند، تا آنجا که ممکن است با خیال راحت برداشته میشود و آنچه باقی مانده است با رادیوسرجری درمان میشود.
تومورهای نخاعی
این تومورها معمولاً مشابه تومورهای هم نوع در مغز درمان میشوند.
آستروسیتومهای نخاع معمولاً نمیتوانند به طور کامل برداشته شوند. آنها ممکن است با جراحی برای برداشتن هر چه بیشتر تومور، به دنبال پرتو درمانی یا تنها با پرتو درمانی درمان شوند. شیمی درمانی ممکن است بعد از جراحی به جای پرتو درمانی در کودکان کوچکتر استفاده شود. در صورتی که به نظر برسد تومور به سرعت در حال رشد است، این روش ممکن است پس از پرتو درمانی در کودکان بزرگتر نیز استفاده شود.
مننژیوم نزدیک نخاع اغلب با جراحی درمان میشود. برخی از اپندیمومها را میتوان با جراحی نیز درمان کرد. اگر اپاندیموم را نتوان به طور کامل برداشت، پرتو درمانی پس از جراحی انجام میشود.
تومورهای شبکه کوروئید (Choroid plexus tumors)
پاپیلومهای خوش خیم شبکه مشیمیه معمولاً فقط با جراحی درمان میشوند. کارسینوم شبکه کوروئید تومورهای بدخیم هستند که فقط گاهی با جراحی درمان میشوند. پس از جراحی، این کارسینومها معمولاً با پرتودرمانی و یا شیمی درمانی درمان میشوند.
کرانیوفارنژیوم (Craniopharyngiomas)
کرانیوفارنژیومها بسیار نزدیک به غده هیپوفیز، اعصاب بینایی و رگهای خونی که مغز را از نظر غذایی تامین میکنند رشد میکنند، بنابراین حذف کامل آنها بدون ایجاد عوارض جانبی دشوار است. برخی از جراحان مغز و اعصاب جراحی را برای برداشتن هر چه بیشتر تومور ترجیح میدهند، در حالی که برخی دیگر ترجیح میدهند بیشتر تومور را برداشته و سپس پرتو درمانی را انجام دهند.
برداشتن جزئی جراحی و به دنبال آن انجام پرتو درمانی بسیار متمرکز ممکن است عوارض جانبی کمتری نسبت به برداشتن کامل تومور ایجاد کند اما هنوز مشخص نیست که آیا این روش در جلوگیری از رشد مجدد تومور خوب است یا خیر.
تومورهای سلولهای زایا
شایع ترین تومور سلول زایا، ژرمینوما (germinoma)، معمولاً با پرتو درمانی به تنهایی قابل درمان است (پس از اینکه با جراحی یا مشاهده نمونه مایع مغزی نخاعی تشخیص داده شد). در صورتی که تومور بسیار بزرگ باشد یا پرتو درمانی آن را به طور کامل از بین نبرد، ممکن است شیمی درمانی به روند درمان اضافه شود.
برخی از پزشکان برای کاهش عوارض جانبی در کودکانی که هنوز به سن بلوغ نرسیده اند، از شیمی درمانی و سپس کاهش دوز پرتو به عنوان درمان اصلی استفاده میکنند. در کودکان بسیار کم سن ممکن است به جای پرتو درمانی از شیمی درمانی استفاده شود. اگر انواع دیگری از تومورهای سلول زایا وجود داشته باشند – خواه مخلوط با ژرمینوما باشند یا نباشند – چشم انداز بیمار معمولاً چندان خوب نیست.
سایر انواع تومورهای سلول زایا (مانند تراتوم و تومور کیسه زرده) به ندرت با جراحی درمان میشوند. برای درمان آنها از پرتو درمانی و شیمی درمانی استفاده میشود اما در برخی موارد ممکن است تومور به طور کامل کنترل نشود. گاهی اوقات این تومورها به مایع مغزی نخاعی (CSF) گسترش مییابند و پرتو درمانی به نخاع و مغز نیز نیاز است.
مطالعات بیشتر در بخش راهنمای علمی سایت
مترجم: فاطمه فریادرس