


بیماری ها
ژنتیک دیابت

ژنتیک دیابت
عوامل خطرزای خانوادگی، پاتوژنز (pathogenesis)، پیگیری های بالینی و درمان دیابت با توجه به علت این بیماری متفاوت است. اگرچه دیابت نوع 2 ریسک خانوادگی بالاتری دارد اما اطلاعات بیشتری در مورد ژنتیک دیابت نوع 1 وجود دارد. ژن هایی که بین 60 تا 65 درصد زمینه ابتلا به دیابت نوع 1 را تشکیل میدهند، شناخته شده اند. دیابت نوع 1 با ژن های زمینه ساز (susceptibility) در ناحیه HLAدر کروموزوم 6p21 و ژن انسولین در کروموزوم 11p15 مرتبط است و حداقل هشت ژن زمینه ساز دیگر نیز تحت بررسی هستند. آنتی بادی های سیتوپلاسمی جزایر لانگرهانس شواهد هومورال خطرساز دیابت نوع 1 را ارائه میدهند. تنها 10 درصد از ژن هایی که زمینه ساز ابتلا به دیابت نوع 2 هستند، شناخته شده اند و عمدتاً با زیرگروه های غیرمعمول این اختلال مرتبط میباشند. ژن گیرنده انسولین روی کروموزوم p1319 و حداقل پنج ژن ناقل گلوکز به ایجاد دیابت نوع 2 کمک میکنند و ممکن است ارتباط های بیشتری از مطالعه ژن گلیکوژن سنتاز، ژن گلوکوکیناز، ژن های MODY و ژن لپتین پدیدار شود. بیماری های ایجاد شده همراه دیابت ممکن است از عوامل زمینه ساز ژنتیکی و محیطی به طور مستقل یا ترکیبی ناشی شوند.
عوامل ژنتیکی در دیابت نوع 1:
دیابت نوع 1 یک اختلال خود ایمنی است که در آن بدن به سلول های بتای پانکراس خود حمله میکند. شروع دیابت نوع 1 هم به یک عامل ارثی و هم به محرک های خارجی مانند رژیم غذایی یا عفونت نسبت داده میشود. جستجو برای شناسایی این عوامل خطرساز ژنتیکی و محیطی ادامه دارد.
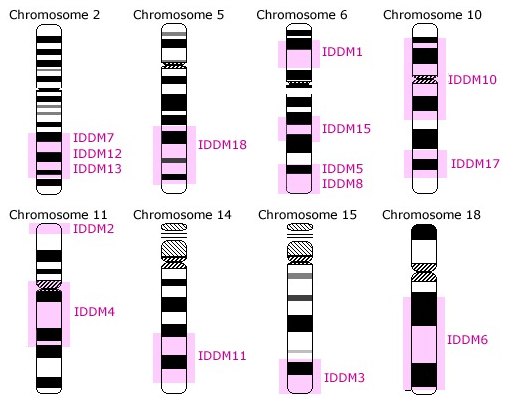
حدود 18 ناحیه از ژنوم با تأثیرگذاری بر خطر دیابت نوع 1 مرتبط است. این مناطق که هر کدام ممکن است حاوی چندین ژن باشند، IDDM1 تا IDDM18 نام گذاری شده اند.
IDDM1 نسبت به همه مناطق دیگر بهتر مورد بررسی و مطالعه قرار گرفته است. این منطقه حاوی ژن های HLA است که پروتئین های دخیل در پاسخ های ایمنی را کد میکند. تغییرات در ژن های HLA یک عامل خطرساز ژنتیکی مهم است اما این تغییرات به تنهایی عامل بیماری نیستند و سایر ژن ها نیز در آن نقش دارند.
دو ژن دیگر نیز غیر از HLA وجود دارند که شناسایی شده اند. یکی از این ژن های غیر HLA (non-HLA)، IDDM2 ژن انسولین است و ژن غیر HLA دیگر، نزدیک به CTLA4 قرار گرفته است که نقش تنظیم کننده ای در پاسخ ایمنی دارد.
IDDM1 حاوی ژن های HLA است:
ژن های HLA مولکول هایی را رمز گذاری میکنند که برای سیستم ایمنی حیاتی هستند. این مولکول ها زنجیره های کوچکی از اسید های آمینه را روی سطح سلول نگه میدارند تا سلول های ایمنی بتوانند این زنجیره ها را تجزیه و تحلیل کرده و در نهایت شناسایی کنند. هنگامی که سلول های ایمنی یک زنجیره نامناسب را پیدا کنند، حمله را آغاز میکنند. بدون ژن های HLA، سلول های ایمنی زنجیره ای از ویروس ها، باکتری ها یا سلول های تومور را پیدا نمیکنند. از سوی دیگر، به ارث بردن نسخه های خاص (آلل) ژن HLA، احتمال حمله سلول های ایمنی به سلول های سالم بدن را افزایش میدهد. این گونه است که IDDM1 به حمله سیستم ایمنی به سلول های بتا و در نتیجه ایجاد دیابت نوع 1 کمک میکند.
ناحیه HLA مجموعه ای از ژن ها در کروموزوم 6 است. ژن ها گلیکوپروتئین هایی را رمز گذاری میکنند که در سطح اکثر سلول ها یافت میشوند و به سیستم ایمنی کمک میکنند تا بین خود (سلولهای خودی، به عنوان مثال، سلولهای بتا پانکراس) و غیر خود (به عنوان مثال، باکتری ها، ویروس ها) تفاوت قائل شود.
بیماری خود ایمنی زمانی ایجاد میشود که سیستم ایمنی به بافت های بدن حمله کند. خطر ابتلا به بیماری های خود ایمنی گاهی به آلل های ژن HLA در بدن مربوط میشود. دیابت نوع 1 در بین این بیماری ها منحصر به فرد است زیرا آلل های HLA چند گونه رفتار کرده و ممکن است خطر ابتلا به دیابت را افزایش دهند، روی آن تأثیری نداشته باشند یا حتی ما را از ابتلا به آن محافظت کنند.
ژنهای HLA پروتئینهایی به نام (MHC) major histocompatibility complex را کد میکنند. دو دسته اصلی از پروتئین های MHC وجود دارد که هر دو زنجیره ای از اسید های آمینه را ارائه میدهند. این زنجیره ها آنتی ژن نامیده میشوند و سلول های ایمنی (به نام سلول هایT) آن ها را تجزیه و تحلیل و شناسایی میکنند. MHC کلاس I زنجیره هایی را از داخل سلول ها ارائه میدهد، در حالی که MHC کلاس 2 زنجیره هایی را از خارج سلول ها ارائه میدهد. اگر سلول های T به زنجیره ارائه شده در MHC متصل شوند، سلول T بلافاصله حملات قدرتمندی را توسط سایر سلول های ایمنی بدن را تدارک میبیند. در حالت ایده آل، بدن فقط حاوی سلول های T است که به زنجیره های ارگانیسم های عفونی (ویروس ها، باکتری ها و غیره) و سلول های تومور متصل میشوند. خلاف این پدیده در بیماری های خود ایمنی مانند دیابت دیده میشود: سلول های T به زنجیره هایی از سلول های سالم بدن متصل شده و آن ها را از بین میبرند.
آلل های مختلفی از ژنهای HLA وجود دارد که منجر به تولید انواع مختلفی از پروتئین های MHC میشوند و اجازه میدهند تا زنجیره های مختلفی توسط سلول ها ارائه شود. وراثت آلل های خاص HLA میتواند بیش از نیمی از ریسک ژنتیکی ابتلا به دیابت نوع 1 را تشکیل دهد. ژن های کد کننده پروتئین های کلاس دو MHC قوی ترین ارتباط را با دیابت دارند. این ژن ها HLA-DR، HLA-DQ و HLA-DP نامیده میشوند.
در جوامع عمومی، تنها نیمی از افراد یک کپی (الل) از ژن DR به نام DR3 و DR4 را به ارث میبرند و کمتر از 3 درصد افراد دارای دو آلل هستند. با این حال، در دیابت نوع 1 حداقل یک آلل DR3 یا DR4 در 95٪ از قفقازی ها یافت میشود و افراد مبتلا به هر دو DR3 و DR4 به طور خاص مستعد ابتلا به دیابت نوع 1 هستند. برعکس، آلل DR2محافظ بوده و مانع ایجاد این بیماری میشود.
مشابه ژن DR، برخی از آلل های ژن DQ نیز از عوامل خطرساز برای ابتلا به این بیماری هستند. در حالی که سایر آلل های DQ محافظ اند. همچنین افرادی که DR3 یا DR4 را به ارث میبرند،DQ را نیز به دارند که خطر ژنتیکی ابتلا به دیابت را افزایش میدهد. برعکس، آلل های محافظ DR و DQ تمایل دارند با هم به ارث برده شوند. این تمایلات مطالعه اثرات ژن های HLA-DR یا HLA-DQ را پیچیده کرده است.
عوامل ژنتیکی در دیابت نوع 2:
دیابت نوع 2 به طور ساده به عنوان دیابت “بزرگسالان” تعریف شده است. اگرچه با شایع تر شدن دیابت در سراسر جهان، موارد دیابت نوع 2 در افراد جوان نیز مشاهده میشود. این نوع از دیابت به طور فزاینده ای در کودکان شایع است.
در تعیین خطر ابتلا به دیابت، عوامل محیطی مانند مصرف غذا و ورزش نقش مهمی دارند. اکثر افراد مبتلا به دیابت نوع 2 دارای اضافه وزن یا چاقی هستند. عوامل ارثی نیز مهم هستند اما ژن های درگیر در این بیماری به خوبی تعریف و شناسایی نشده اند.
در اشکال نادر دیابت، جهش در یک ژن میتواند منجر به بیماری شود. با این حال تصور میشود که در دیابت نوع 2 ژن های زیادی دخیل هستند. “ژن های دیابت” ممکن است تنها یک تغییر ظریف در توالی ژن را نشان دهند و این تغییرات ممکن است بسیار رایج باشند. سختی این مطالعات در ربط دادن چنین واریانت های ژنی رایجی، معروف به پلیمورفیسم های تک نوکلئوتیدی (SNPs)، با افزایش خطر ابتلا به دیابت است.
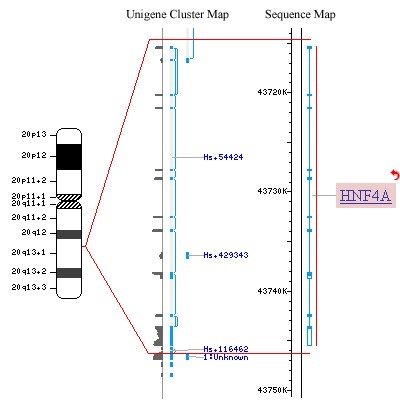
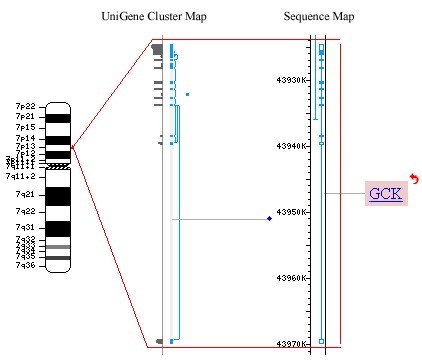
یکی از روش های یافتن ژن های زمینه ساز دیابت، مطالعه ارتباط کل ژنوم (whole-genome linkage) است. در این روش کل ژنوم اعضای خانواده مبتلا اسکن شده و خانواده ها در طول چندین نسل بررسی میشوند و یا تعداد زیادی از جفت خواهر و برادران مبتلا، مورد مطالعه قرار میگیرند. سپس ارتباط بین بخش هایی از ژنوم و خطر ابتلا به دیابت بررسی میشود. تا به امروز تنها دو ژن، کالپین 10 (CAPN10) و فاکتور هسته ای هپاتوسیت 4 آلفا (HNF4A) با این روش شناسایی شده اند.
گیرنده سولفونیل اوره (ABCC8):
سولفونیل اوره ها دسته ای از دارو هایی هستند که برای کاهش گلوکز خون در درمان دیابت نوع 2 استفاده میشوند. این دارو ها به گیرنده سولفونیل اوره سلول های بتای پانکراس متصل شده و ترشح انسولین را تحریک میکنند. گیرنده سولفونیل اوره توسط ژن ABCC8 کد گذاری میشود. تنوع ژنتیکی در ژن ABCC8 ممکن است آزاد سازی انسولین را مختل کند.
پروتئین کد گذاری شده توسط ژن ABCC8 عضوی از انتقال دهنده های کاست (cassette) متصل به ATP است. این پروتئین ها از انرژی به شکل ATP برای هدایت انتقال مولکول های مختلف در غشای سلولی استفاده میکنند. ABCC8 متعلق به زیرخانواده ای از ناقلین است که حاوی کانال کلریدی – که در فیبروز کیستیک (CFTR) دچار جهش شده است – و همچنین پروتئین هایی که در مقاومت چند دارویی (multi-drug resistance) دخیل هستند، میباشد.
پروتئین ABCC8 به عنوان گیرنده سولفونیل اوره (SUR) نیز شناخته میشود. SUR یکی از پروتئین هایی است که کانال پتاسیم حساس به ATP (KATP) موجود در پانکراس را ایجاد میکند. پروتئین دیگری به نام Kir6.2 هسته کانال را تشکیل میدهد که توسط ژن KCNJ11 کدگذاری میشود. کانالهای KATP با پیوند دادن سیگنال های حاصل از متابولیسم گلوکز (افزایش ATP) به دپلاریزاسیون غشا (به دلیل بسته شدن کانالهایKATP ) و ترشح انسولین، نقش اصلی را در ترشح انسولین ناشی از گلوکز بازی میکنند.
فعالیت کانال KATP ترشح انسولین را تنظیم میکند. سولفونیل اوره ها داروهایی هستند که میتوانند فعالیت کانال KATP را تعدیل کنند و در درمان دیابت نوع 2 استفاده میشوند. آن ها با اتصال به SUR کانال را مهار کرده و ترشح انسولین را تحریک میکنند. این پدیده منجر به کاهش سطح گلوکز خون میشود.
فعالیت کانال KATP نیز توسط نوع فرعی دیگری از SUR تعدیل می شود. (SUR که همچنین به عنوان SUR1 شناخته شده و توسط ABCC8 کدگذاری می شود؛ یا SUR2A و SUR2B که توسط ABCC9 کدگذاری میشوند.) تصور می شود که در لوزالمعده، اکثر کانال های KATP مجموعه ای از چهار پروتئین SUR1 و چهار پروتئین Kir6.2 هستند.
جهش در ABCC8 یا KCNJ11 میتواند منجر به افزایش ترشح انسولین شود؛ وضعیتی که هیپوگلیسمی هایپرانسولینمی خانوادگی پایدار در دوران نوزادی (PHHI) نامیده میشود. تنوع ژنتیکی در ABCC8 نیز در اختلال در ترشح انسولین که در دیابت نوع 2 دیده میشود، دخیل است.
دو پلی مورفیسم رایج ژن ABCC8 (اگزون 16-3t/cو اگزون 18 T/C) با دیابت نوع 2 ارتباط متغیری دارند. با این حال، اخیراً یک مطالعه بزرگ در بریتانیا نشان داد که شاید این گونه های ABCC8 با دیابت مرتبط نباشند.
انواع دیگر دیابت:
اکثریت قریب به اتفاق موارد دیابت در دسته های دیابت نوع 1، نوع 2 و دیابت دوران بارداری (gestational diabetes) قرار میگیرند. با این حال، بالای 5 درصد از موارد، دلایل خاص دیگری دارند و مبتلا به دیابتی هستند که از جهش در یک ژن منفرد ناشی میشود.
نقص ژنتیکی در عملکرد سلول های بتا ( MODYو سایر موارد):
- MODY
(MODY) Maturity onset diabetes in the young یک علت غیر معمول ابتلا به دیابت است که ممکن است با دیابت نوع 2 اشتباه گرفته شود زیرا درمان هر دو این بیماری ها – حداقل در مراحل اولیه – نیازی به انسولین ندارند.
با این حال، تفاوت های زیادی بین MODY و دیابت نوع 2 وجود دارد. در حالی که ماهیت پتانسیل ژنتیکی دیابت نوع 2 با وجود بسیاری از ژن های زمینه ای مشخص نیست، MODY یک وضعیت تک ژنی است (ناشی از جهش یک ژن منفرد) که به عنوان یک صفت غالب اتوزومی به ارث میرسد. شروع دیابت معمولاً در دوران کودکی یا نوجوانی و معمولاً قبل از 25 سالگی رخ میدهد. اگرچه هایپرگلیسمی در برخی موارد خفیف است و ممکن است مانند دیابت نوع 2 در فرد نمایان نشود. هنگامی که هایپرگلیسمی در کودکان دیده شود، ممکن است MODY به اشتباه به عنوان دیابت نوع 1 تشخیص داده شود.
مطالعات ژنتیکی تعدادی زیرگروه MODY را تعریف کرده است. جهش در ژن های کد کننده فاکتور هسته ای 4 کبدی (HNF4)، گلوکوکیناز (GCK)، فاکتور هسته ای 1 آلفا و 1 بتا (که معمولاً به عنوان HNF1A و HNF1B شناخته میشوند اما نماد های رسمی آن ها به ترتیب TCF1 و TCF2میباشد)، فاکتور پروموتور انسولین 1 (IPF-1) و NEUROD1 علت شش شکل شناخته شده این نوع(MODY1-6) هستند.
MODY2 توسط یک آنزیم گلوکوکیناز جهش یافته ایجاد میشود که نمیتواند غلظت گلوکز در گردش را به طور دقیق تشخیص دهد. تمام ژن های باقی مانده MODY فاکتور های رونویسی را رمزگذاری میکنند. HNF4A، TCF1،TCF2 و IPF-1 پیوند های مهمی را در زنجیره فاکتور های رونویسی تشکیل میدهند که بیان مناسب ژن های سلول های بتا، مانند انسولین و ناقل گلوکز GLUT2 را کنترل میکنند. جهش در این ژن ها ممکن است رشد سلول های بتا را در جنین مختل کند و منجر به اختلال در عملکرد سلول های بتا در بزرگسالان شود. با این حال، نقش دقیق این پروتئین ها در جزایر لانگرهانس پانکراس بزرگسالان، تازه در حال آشکار شدن است.
MODY3 و MODY2 شایع ترین علل MODY هستند اما همچنان علل نسبتاً غیر معمولی برای دیابت باقی میمانند.
سایر جهش هایی که با اختلال در عملکرد سلول های بتا باعث دیابت میشوند:
جهش در DNA میتوکندری یکی از علل نادر دیابت است. DNA میتوکندری یک مولکول دایره ای بوده که شامل 37 ژن است که از مادر به فرزندانش منتقل میشود. تصور میشود که انتقال DNA میتوکندری از پدر انجام نمیشود زیرا پس از لقاح، تخم بارور شده میتوکندری های حاصل از اسپرم را از بین میبرد.
دیابت و کم شنوایی با جهش نقطه ای DNA میتوکندریایی همراه است. این جهش در ژنی که لوسین tRNA را کد میکند، رخ میدهد و منجر به جایگزینی گوانین به جای آدنین (A→G) در موقعیت 3243 میشود.
جهش tRNA Leu 3243 در ابتدا در بیماران مبتلا به سندرم MELAS (میوپاتی میتوکندری، آنسفالوپاتی، اسیدوز لاکتیک و سندرم شبه سکته مغزی) شناسایی شد. با این حال، دیابت بخشی از این سندرم نیست و این موضوع نشان میدهد که این جهش میتوکندریایی ممکن است به صورت فنوتیپ های مختلف بیان شود.
MODY1: ناشی از جهش در فاکتور رونویسی HNF4A:
ژن HNF4A یک فاکتور رونویسی را رمزگذاری میکند که در کبد و پانکراس یافت میشود. HNF4A در ناحیه ای از کروموزوم 20 یافت میشود که با دیابت نوع 2 مرتبط است و جهش های این ژن، شکل نادری از دیابت اتوزومال غالب (MODY1) را بوجود میآورد.
بیان طیف وسیعی از ژن ها در کبد توسط فاکتور رونویسی هسته ای هپاتوسیت 4 آلفا (HNF4A) تنظیم میشود. بسیاری از عملکرد هایی که کبد انجام میدهد بسته به اینکه آیا HNF4A بیان میشود یا نمیشود، ممکن است رخ دهند یا از بین بروند. علاوه بر این، HNF4A بیان یک فاکتور رونویسی دیگر، فاکتور هسته ای هپاتوسیت 1 آلفا (HNF1A) را کنترل میکند که به نوبه خود تنظیم بیان چندین ژن مهم در کبد از جمله HNF4A را بر عهده دارد.
همان طور که از نام آن پیداست، HNF4A به وفور در کبد یافت شده اما در سلول های بتا پانکراس، کلیه ها و روده ها نیز وجود دارد. آن ها همراه با سایر فاکتور های رونویسی مانند HNF1A و HNF1B (به ترتیب توسط TCF1 و TCF2 کد گذاری میشوند)، بخشی از شبکه ای از فاکتور های رونویسی را تشکیل میدهند که با هم برای کنترل بیان ژن در جنین در حال رشد همکاری میکنند. به طور خاص، تصور میشود که HNF4A نقش مهمی در رشد کبد، کلیه و روده دارد.
در سلول های بتای پانکراس، این شبکه فاکتور های رونویسی بیان ژن انسولین را تنظیم میکند. علاوه بر این،HNF4 و HNF1 بیان چندین ژن دیگر مرتبط با ترشح انسولین را تنظیم میکنند؛ به عنوان مثال، ژن هایی که پروتئین های دخیل در انتقال گلوکز (GLUT2) و متابولیسم گلوکز و میتوکندری را کد میکنند.
به نظر میرسد که ژن HNF4A در دیابت نوع 2 نقش داشته باشد. ارث بردن انواع خاصی از HNF4A ممکن است ترشح انسولین را تغییر داده و مستعد ایجاد هایپرگلیسمی باشد. جهش HNF4A میتواند باعث ایجاد یک نوع بسیار نادر از دیابت (MODY1) شود. در حالی که دیابت نوع 2 اختلالی است که معمولاً دیر ظاهر میشود و اساس پلی ژنتیکی قابل توجهی دارد،MODY طبق آمار در جوانان (در سن کمتر از 25 سال) شروع میشود و یک اختلال تک ژنتیکی است که به روش اتوزومال غالب به ارث میرسد.
MODY2: ناشی از جهش در آنزیم گلوکوکیناز :(GCK)
گلوکوکیناز که توسط ژن GCK کد گذاری میشود، اولین مرحله متابولیسم گلوکز را در کبد کاتالیز میکند. همچنین ممکن است یک “حسگر گلوکز” مهم در پانکراس باشد. گلوکوکیناز جهش یافته باعث ایجاد یک فرم نادر اتوزومال غالب دیابت (MODY2) شده و ممکن است در دیابت نوع 2 نیز نقش داشته باشد.
آنزیم گلوکوکیناز متابولیسم گلوکز را در کبد و سلول بتا پانکراس کاتالیز میکند. گلوکوکیناز، گلوکز را در داخل سلول با کاتالیز کردن فسفوریلاسیون آن، برای تولید گلوکز-6-فسفات به دام میاندازد. این اولین مرحله و محدود کننده سرعت در گلیکوز است، مسیری که انرژی را به شکل ATP از گلوکز تولید میکند.
گلوکوکیناز (همچنین هگزوکیناز IV نامیده میشود) با سایر هگزوکیناز هایی که در بافت های دیگر یافت میشوند، متفاوت است. اول این که گلوکوکیناز تمایل کمتری به گلوکز دارد. این پدیده به سایر اندام ها مانند مغز و ماهیچه ها اجازه میدهد تا زمانی که عرضه هگزوکیناز ها محدود است، گلوکز را دریافت و جذب کنند. ویژگی دوم این است که گلوکوکیناز توسط محصول آن، گلوکز-6-فسفات مهار نمیشود. این پدیده عدم مهار بازخورد منفی، گلوکوکیناز کبدی را قادر میسازد تا زمانی که گلوکز فراوان است، فعال باقی بماند و مطمئن شود که کبد میتواند کل گلوکز اضافی را از خون حذف کرده و هیچ گلوکزی را هدر نمیدهد.
گلوکوکیناز به عنوان یک “حسگر گلوکز” مهم در فرآیند زیر عمل میکند. سرعت متابولیسم گلوکز با سرعت فسفوریلاسیون گلوکز تعیین میشود که توسط گلوکوکیناز در کبد و پانکراس کاتالیز میگردد. کبد و لوزالمعده همچنین انتقال دهنده گلوکز-2 (GLUT2) را بیان میکنند – یک پروتئین سلولی مستقل از انسولین که واسطه انتقال گلوکز به سلول ها است. ظرفیت GLUT2 برای انتقال گلوکز بسیار بالا است و سریعاً تعادل بین گلوکز خارج سلولی و درون سلولی را تسهیل میکند. بنابراین، در واقع، غلظت گلوکز خارج سلولی توسط گلوکوکیناز حس میشود.
گلوکوکیناز با کاتالیز کردن مرحله محدود کننده سرعت متابولیسم گلوکز در کبد، کبد را قادر میسازد تا افزایش گلوکز را که بعد از خوردن غذا اتفاق میافتد، مهار کند. در پانکراس، گلوکوکیناز حسگر گلوکز برای ترشح انسولین است. آستانه و حد آزاد سازی انسولین تحریک شده با گلوکز، حدود 5 میلی مول در لیتر است. جهش های GCKکه این آستانه را تغییر میدهند به عنوان سه سندرم مختلف ظاهر میشوند و اهمیت GCK را در هموستاز گلوکز و دیابت برجسته میکنند:
سندروم اول:
جهش های فعال، آستانه سلول های بتا را برای آزاد سازی انسولین به 1.5 میلی مول در لیتر گلوکز کاهش میدهند که منجر به افزایش ترشح انسولین میشود. این پدیده به صورت هایپوگلیسمی هیپرانسولینمی مداوم در دوران نوزادی (PHHI) ظاهر میشود.
سندروم دوم:
جهش های غیر فعال، آستانه سلول های بتا را برای آزاد سازی انسولین افزایش میدهند. اگر دو آلل تغییر یافته توسط جهش های غیر فعال به ارث برده شوند، سطح گلوکز مورد نیاز برای تحریک ترشح انسولین از پانکراس بسیار بالا است. این افراد از بدو تولد مبتلا به نوعی دیابت به نام دیابت نوزادی دائمی (permanent neonatal diabetes) هستند.
سندروم سوم:
MODY2 به دلیل به ارث بردن یک آلل است که توسط یک جهش غیر فعال تغییر یافته است. این غیر فعال سازی جزئی، منجر به افزایش ترشح انسولین تحریک شده با گلوکز، به حدود 7 میلی مول در لیتر میشود. این پدیده یک هایپرگلیسمی خفیف را در فرد ایجاد میکند که از بدو تولد وجود دارد اما اغلب فقط در اواخر زندگی تشخیص داده میشود.
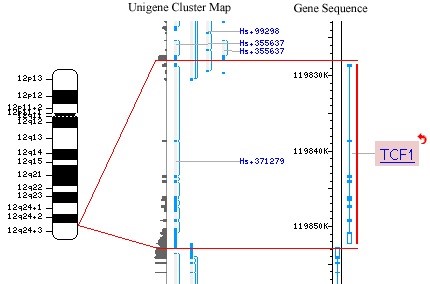
MODY3: ناشی از جهش در فاکتور رونویسی TCF1
ژن TCF1 یک فاکتور رونویسی را کد میکند که در کبد و پانکراس یافت میشود و در رشد این اندام ها و همچنین سایر اندام ها پر اهمیت است. جهش TCF1 باعث ایجاد شکل نادری از دیابت اتوزومال غالب (MODY3) میشود.
TCF1 متعلق به شبکه ای از فاکتور های رونویسی است که بیان طیف وسیعی از ژن ها را در کبد هماهنگ میکند. در جنین، این شبکه از پروتئین های هسته ای، رشد کبد را هدایت میکنند و در بزرگسالان نیز همچنان مهم اند. بسیاری از عملکرد های کبد بسته به بیان این عوامل رونویسی ظاهر و یا ناپدید میشوند.
اگرچه این شبکه از فاکتور های رونویسی در بالاترین مقدار خود در کبد یافت می شود اما در سایر اندام ها مانند پانکراس و کلیه نیز وجود دارد. فاکتور رونویسی HNF4A بیان TCF1 را تنظیم میکند و ممکن است در تنظیم بیان ژن TCF2 نیز نقش داشته باشد.
در سلول های بتای پانکراس،HNF4 و TCF1 بیان ژن انسولین را به همراه چندین ژن دیگر مرتبط با ترشح انسولین تنظیم میکنند. به عنوان مثال، ژن هایی که پروتئین های دخیل در انتقال گلوکز (GLUT2) و متابولیسم گلوکز را کد میکنند. از آن جایی که مشخص شده استTCF2 در جزایر لانگرهانس پانکراس بیان میشود، گمان میرود که TCF2 با TCF1 برای تنظیم بیان ژن در سلول های بتا عمل میکنند.
TCF1 و TCF2 دمین (domains) های مشابهی دارند. آن ها دارای یک منطقه اتصال DNA و دمین دیمریزاسیون مشابه هستند. اعتقاد بر این است که پروتئین TCF2 با TCF1 هترودایمر ها (heterodimers) را تشکیل میدهند و بسته به ایزوفرم TCF2، نتیجه ممکن است فعال یا مهار کردن رونویسی ژن های هدف باشد.
جهش های HNF4A، TCF1 و TCF2 هر کدام باعث ایجاد شکل مشخصی از دیابت MODY میشوند. جهش HNF4A باعث MODY1، جهش TCF1 باعث MODY3 و جهش TCF2 باعث MODY5 میشود.
MODY4: ناشی از جهش در فاکتور رونویسی IPF1
فاکتور پروموتور انسولین ۱ (IPF1) مسئول رشد پانکراس در جنین بوده و همچنین تنظیم کننده کلیدی بیان ژن انسولین است. جهش IPF1 باعث ایجاد شکل نادری از دیابت اتوزومال غالب (MODY4) شده و ممکن است در احتمال ابتلا به دیابت نوع 2 نیز نقش داشته باشد.
در جنین در حال رشد، لوزالمعده از دو جوانه روده اولیه تشکیل میشود که در نهایت با هم ادغام شده و غده پانکراس را تشکیل میدهند. بخش برون ریز پانکراس شامل سلول هایی است که آنزیم های گوارشی مانند پروتئاز ها و لیپاز ها را تولید میکنند که از طریق مجاری پانکراس به روده ترشح میشوند. غدد درون ریز پانکراس بسیار کوچک تر بوده و عمدتاً از سه نوع سلول – آلفا، بتا و دلتا – تشکیل شده اند که به ترتیب گلوکاگون، انسولین و سوماتوستاتین (somatostatin) را تولید میکنند.
رشد پانکراس به خوبی مورد مطالعه قرار گرفته است و بسیاری از فاکتور های رونویسی را می توان برای شناسایی سلول های پانکراس در مراحل مختلف رشد استفاده کرد. فاکتور 1 پروموتور انسولین (IPF1) یکی از این فاکتور های رونویسی بوده و یک نشانگر اولیه پانکراس است که در سلول های بتا بالغ نیز یافت میشود. کمی بعد، در مراحل رشد پانکراس، TCF1 بیان شده و همچنین در سلول های بتا بالغ نیز یافت میشود.
در جنین، وجود IPF1 برای اطمینان از رشد صحیح پانکراس حیاتی است. از دست دادن هر دو نسخه از این ژن میتواند باعث تشکیل نشدن پانکراس شود (pancreas agenesis). بدونIPF1، تکثیر و تمایز سلول های پیش ساز به قسمت های غدد درون ریز و برون ریز پانکراس مسدود شده و این فرآیند مختل میشود.
IPF1 همچنان برای عملکرد طبیعی پانکراس در بزرگسالان ضروری است. IPF1 بیان چندین ژن پانکراس را تنظیم میکند که مهم ترین آن ها ژن انسولین (INS)، ناقل گلوکز نوع 2 (GLUT2)، گلوکوکیناز (GCK) و سوماتوستاتین است. از دست دادن یک نسخه از IPF1 با MODY4 مرتبط است و ممکن است در احتمال ابتلا به دیابت نوع 2 نقش داشته باشد.
نقش دوگانه IPF1 در طول رشد پانکراس در جنین و به عنوان یک تنظیم کننده برای ژن های پانکراس در بزرگسالان بر اهمیت IPF1 در هموستاز گلوکز تأکید میکند.
MODY5: ناشی از جهش در فاکتور رونویسی TCF2
TCF2 یک فاکتور رونویسی را رمز گذاری میکند که در کبد و پانکراس یافت میشود و در رشد این اندام ها و همچنین سایر اندام ها حائز اهمیت است. جهش TCF2 باعث شکل نادری از دیابت اتوزومال غالب (MODY5) میشود.
TCF2 متعلق به شبکه ای از فاکتور های رونویسی است که بیان طیف وسیعی از ژن ها را در کبد هماهنگ میکند. در جنین، این شبکه از پروتئین های هسته ای، رشد کبد را هدایت میکند و در بزرگسالان نیز همچنان پر اهمیت اند. بسیاری از عملکرد های کبد بسته به بیان این عوامل رونویسی ظاهر و ناپدید میشوند.
اگرچه این شبکه از فاکتور های رونویسی در بالاترین مقدار در کبد یافت میشود اما در سایر اندام ها مانند پانکراس و کلیه نیز حضور دارد. فاکتور رونویسی HNF4A بیان TCF1 را تنظیم کرده و ممکن است در بیان ژن TCF2 نیز موثر باشد.
در سلول های بتای پانکراس، HNF4 و TCF1 بیان ژن انسولین را به همراه چندین ژن دیگر مرتبط با ترشح انسولین تنظیم میکنند. به عنوان مثال، ژن هایی که پروتئین های دخیل در انتقال گلوکز و متابولیسم گلوکز را کد میکنند. از آنجا که TCF2 در جزایر لانگرهانس پانکراس بیان میشود، به نظر میرسد که TCF2 با TCF1 برای تنظیم بیان ژن در سلول های بتا همکاری میکند.
TCF1 و TCF2 دمین های مشابهی دارند. آن ها دارای یک منطقه اتصال DNA و دمین دیمریزاسیون مشابه هستند. اعتقاد بر این است که پروتئین TCF2 با TCF1 هترودایمر ها را تشکیل میدهند و بسته به ایزوفرم TCF2، نتیجه ممکن است فعال یا مهار کردن رونویسی ژن های هدف باشد.
جهش های HNF4A،TCF1 و TCF2 هر کدام باعث ایجاد شکل مشخصی از MODY میشوند. جهش HNF4A باعث MODY1، جهش TCF1 باعث MODY3 و جهش TCF2 باعث MODY5 میشود.
MODY6: ناشی از جهش در فاکتور رونویسی NEUROD1
فاکتور رونویسی NEUROD1 میتواند مستقیماً رونویسی ژن انسولین را فعال کند. همچنین در رشد سلول های بتای پانکراس و سیستم عصبی مورد نیاز است. جهش های این ژن باعث ایجاد MODY6 میشود که جدیدترین شکل کشف شده از دیابت اتوزومال غالب است و ممکن است در دیابت نوع 1 و 2 نیز نقش داشته باشد.
توسعه و عملکرد طبیعی غدد درون ریز پانکراس وابسته به شبکه ای از عوامل رونویسی است. این پروتئین ها به صورت منفی یا مثبت بر رونویسی ژن ها تأثیر میگذارند. ژن NEUROD1 یک فاکتور رونویسی را کد میکند که تنظیم کننده مثبتی برای رونویسی ژن انسولین است.
NEUROD1 (برای “تمایز نوروژنیک”) پروتئینی است که اولین بار برای نقش پر اهمیت آن در رشد سیستم عصبی جنینی کشف شد. بیان NEUROD1 نورون ها را به بلوغ یا تمایز تحریک میکند و این پتانسیل را دارد که سلول های تمایز نیافته را به نورون ها تبدیل کند.
در مدل های حیوانی، جهش های ژن NEUROD1 رشد طبیعی پانکراس را مختل کرده و منجر به ایجاد دیابت میشود.
در اثر این جهش ساختار های خاصی در مغز نیز، مانند مخچه و هیپوکامپ به درستی رشد نمیکنند و در نتیجه تشنج ایجاد میشود.
ارتباط بین NEUROD1 و دیابت اولین بار زمانی مطرح شد که دانشمندان کشف کردند ژن NEUROD1 در همان ناحیه از کروموزوم قرار دارد که با پتانسیل ابتلا به دیابت نوع 1 مرتبط است. این ناحیه IDDM7 نام داشته و روی بازوی کوتاه کروموزوم 2 قرار دارد.
علاوه بر ارتباط با دیابت نوع 1، انواع NEUROD1 نیز با احتمال ابتلا به دیابت نوع 2 مرتبط اند و جهش NEUROD1 باعث ایجاد جدید ترین شکل کشف شده دیابت اتوزومال غالب، MODY6 میشود.
مترجم: فاطمه فریادرس
مطالعه صدها مطلب علمی در حوزه بیولوژی
آرشیو جدیدترین خبرهای روز دنیای بیولوژی