


اطلاعات عمومی، ویکی ژن
FASTA: تعریف، برنامهها، طرز کار، مراحل و کاربردها
مقدمهای بر ابزار FASTA
پایگاه داده جستجوی شباهت یک تکنیک ضروری در بیوانفورماتیک (Bioinformatic) است زیرا به ما امکان میدهد توالیهای تازه تعیین شده را با مقایسه آنها با پایگاههای داده موجود توصیف کنیم.
FASTA یکی از اولین ابزارهای جستجوی شباهت در پایگاه داده پرکاربرد است. FASTA (یا FastA)، مخفف «Fast-All»، ابزاری برای همترازسازی توالی (Sequence alignment) است که توالیهای نوکلئوتیدی یا پروتئینی را به عنوان ورودی میگیرد و آن را با پایگاههای داده موجود مقایسه میکند. اولین بار توسط David J. Lipman و William R. Pearson در سال 1985 توسعه یافت و از آن زمان تا کنون برای کاربردهای مختلف اصلاح و تعدیل شده است.
فرمت فایل مبتنی بر متن (Text-based file format) برای نمایش توالیهای نوکلئوتیدی یا پروتئینی که از برنامه FASTA نشأت میگیرد، اکنون به یک استاندارد در بیوانفورماتیک تبدیل شده است. بسیاری از ابزارهای جستجوی توالی در پایگاه داده دیگر نیز از فرمت فایل FASTA استفاده میکنند.

برنامههای FASTA
FASTA در ابتدا برای مقایسه توالی پروتئین توسعه داده شد. برنامه اصلی به عنوان FASTP نامیده میشد. این برنامه به سرعت به ابزاری محبوب برای همترازسازی توالی و جستجو در پایگاه داده تبدیل شد. FASTA به طور مداوم به روز شده و بهبود یافته است. در حال حاضر برنامههای مختلف FASTA در دسترس هستند که هر کدام برای انواع متفاوتی از جستجوهای دنباله استفاده میشوند:
- FASTA با استفاده از الگوریتم FASTA، یک توالی مورد بررسی DNA را با یک پایگاه داده از توالیهای DNA یا یک توالی مورد بررسی پروتئین را با پایگاه دادهای از توالیهای پروتئینی مقایسه میکند.
- SSEARCH با استفاده از الگوریتم Smith-Waterman، مقایسه پروتئین-پروتئین یا DNA-DNA را انجام میدهد.
- GGSEARCH/GLSEARCH با استفاده از یک الگوریتم همترازسازی سراسری (GGSEARCH) یا ترکیبی از الگوریتمهای همترازسازی سراسری و محلی (GLSEARCH) به مقایسه توالیهای پروتئینی و نوکلئوتیدی میپردازد.
- FASTX/FASTY یک توالی DNA و یک پایگاه داده از توالیهای پروتئین را با ترجمه توالی DNA به سه فریم و اجازه دادن به شکافها و جهشهای تغییر چارچوب (frameshift)، مقایسه میکند.
- TFASTX/TFASTY یک توالی پروتئین و یک پایگاه داده از توالیهای DNA را مقایسه میکند. توالی DNA در شش فریم ترجمه میشود: سه فریم در جهت جلو و سه فریم در جهت معکوس.
- FASTF/TFASTF توالیهای پپتیدی (Peptide) مخلوط را با پایگاه دادههای پروتئین (FASTF) یا DNA ترجمه شده (TFASTF) مقایسه میکند.
- FASTS/TFASTS مجموعهای از قطعات پپتیدی کوتاه را با پایگاه دادههای پروتئین (FASTS) یا DNA ترجمه شده (TFASTS) مقایسه میکند.
FASTA چگونه کار میکند
FASTA با مقایسه یک دنباله مورد بررسی با پایگاه دادهای از توالیها برای شناسایی مطابقتهای مشابه کار میکند. این برنامه از یک الگوریتم ابتکاری (Heuristic algorithm) برای جستجوی سریع پایگاه داده و شناسایی مهمترین تطابقهای مشابه استفاده میکند.
مکانیسم کار FASTA در مراحل زیر شرح داده شده است:
مرحله 1: شناسایی بخشها
اولین مرحله شناسایی بخشها با شباهت بالا با ایجاد یک لوکاپ تیبل (Lookup table) برای دنباله مورد بررسی است. به این مرحله مرحله هشینگ (hashing) نیز میگویند. برای ایجاد لوکاپ تیبل، ابتدا دنباله مورد بررسی به متنهای کوچکتر معروف به k-tuple (ktup) تقسیم میشود.
هنگامی که مقدار ktup افزایش مییابد، تعداد بازدیدهای متن پسزمینه کاهش مییابد. با کاهش تعداد بازدیدهای این متنهای پسزمینه، الگوریتم میتواند روی بازدیدهای مرتبطتر تمرکز کند و سرعت کلی جستجو را افزایش دهد. k-tuple معمولا برای پروتئینها برابر 2 و برای توالیهای نوکلئوتیدی برابر 6 است.
هنگامی که لوکاپ تیبل ایجاد شد، برای شناسایی تطابق بین k-tupleها در دنباله مورد بررسی و توالیها در پایگاه داده استفاده میشود. بخشهای مشابه به صورت قطری در یک ماتریس دو بعدی نشان داده میشوند. ده بخش با بیشترین تراکم تطابق متنها، مناطق با شباهت بالا هستند و بهترین ده قطر ذخیره میشوند.
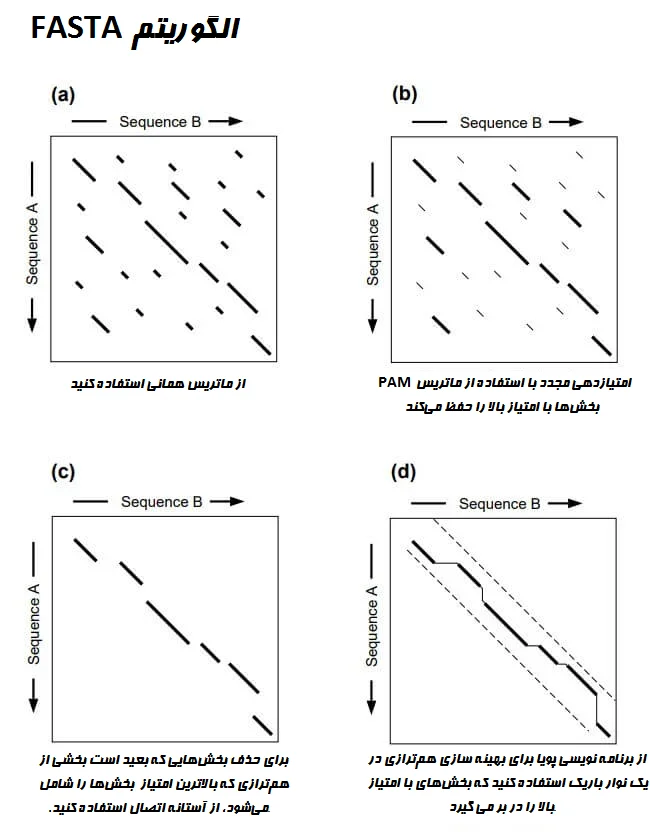
مرحله 2: امتیازدهی مجدد
در مرحله دوم ده قطر برتر با استفاده از ماتریسهای امتیازدهی مناسب، مجدداً امتیازدهی میشوند. برای پروتئین، ماتریس BLOSUM50 یا PAM استفاده میشود. برای توالیهای DNA از ماتریس همانی (Identity matrix) استفاده میشود. یک زیربخش با بالاترین امتیاز برای هر یک از بخشهای قطری اسکن شده، شناسایی میشود. به این زیربخشها با امتیاز بالا در قطرها، بخشهای اولیه میگویند.
مرحله 3: آستانه (Threshold) اتصال
در مرحله بعد، یک برش امتیاز یا آستانه اتصال اعمال میشود که بخشهایی را که بعید است قسمتی از همترازسازی نهایی باشند، حذف میکند. توالیهای کتابخانه بر اساس امتیازات اولیه آنها رتبهبندی میشوند.
بخشهایی که امتیازات اولیه آنها بالاتر از ترشهلد از پیش تعیین شده است، انتخاب شده و بررسی میشوند تا ببینیم آیا میتوان آنها را به یکدیگر متصل کرد یا خیر. این مرحله در حین اعمال جریمههای شکاف، شکافهایی را بین قطرها معرفی میکند. امتیاز همترازی شکافدار با کم کردن یک جریمه برای هر شکاف محاسبه میشود که برای رتبهبندی توالیها در پایگاه داده بر اساس شباهت استفاده میشود.
مرحله 4: همترازی نهایی
در نهایت، همترازی شکافدار برای تولید همترازسازی اصلاح میشود. این کار با استفاده از الگوریتم Smith-Waterman نواری انجام میشود. این الگوریتم یک الگوریتم برنامهنویسی پویا بوده که امتیاز بهینه (opt) را برای همترازسازی محاسبه میکند. این امتیاز برای محاسبات آماری استفاده میشود.
معناداری آماری و FASTA
FASTA همچنین تخمینی از معناداری آماری (Statistical Significance) همترازی یافت شده ارائه میدهد. این معنیداری با استفاده از امید ریاضی (E-value) که احتمال به دست آوردن امتیاز همترازی توالی به طور تصادفی را اندازهگیری میکند، ارزیابی میشود. هر چه امید ریاضی کوچکتر باشد، هم ترازی معنادارتر است.
E-value تنها پارامتر آماری نیست. FASTA همچنین از معیارهای آماری دیگری مانند امتیاز بیت (Bit score) و امتیاز شباهت (Similarity score) بر اساس ماتریس امتیازدهی و جریمههای شکاف برای ارزیابی معناداری همترازیهای توالی استفاده میکند.
خروجی FASTA همچنین شامل یک پارامتر آماری اضافی به نام Z-score است که تعداد انحرافات استاندارد از میانگین امتیاز جستجو در پایگاه داده را نشان میدهد. یک مقدار Z بالاتر نشان دهنده تطابق معنادارتر است.
کاربردهای FASTA
FASTA طیف وسیعی از کاربردها دارد که برخی از آنها عبارتند از:
- FASTA میتواند در همترازسازی توالی برای شناسایی بخشهای مشابه استفاده شود. FASTA در شناسایی بخشهای محفاظت شده در توالیهای DNA یا پروتئین مفید است، که میتواند به شناسایی حوزهها یا موتیفهای (Motif) کارا کمک کند. شناسایی این حوزهها یا موتیفهای کارا میتواند بینشهایی در مورد کارکرد بیولوژیکی دنباله ارائه دهد.
- از FASTA میتوان برای جستجو در پایگاه دادههای بزرگ از توالیها برای یافتن مطابقت با یک دنباله مورد بررسی استفاده کرد. FASTA به شناسایی توالیهای همولوگ (Homologous) کمک میکند که میتواند به پیشبینی کارکرد یک دنباله تازه شناسایی شده، کمک کند.
- FASTA میتواند درختان فیلوژنتیک (Phylogenetic tree) را با تراز کردن توالی از گونههای مختلف و شناسایی روابط تکاملی بین آنها، بسازد.
همچنین بخوانید:
- آنالیز فیلوژنتیک
- زبان برنامهنویسی پایتون و R در بیوانفورماتیک
- مدلسازی همسانی (Homology Modeling) – سازوکار، مراحل و کاربردها
- طراحی پرایمر چیست؟
مترجم: صادق حسینیکیا