



مقدمهای بر سندرم Singleton-Merten
سندرم Singleton-Merten یک اختلال بسیار نادر است که ویژگیهای اصلی آن شامل ناهنجاریهایی در دندانهای فرد بیمار (Dental dysplasia)، کلسیفیکاسیون (Calcifications) در آئورت و دریچههای قلب (به عنوان مثال، دریچههای آئورت و میترال) است. همچنین نازک شدن استخوانها و از بین رفتن تدریجی بافت استخوانها (osteoporosis) در این افراد دیده میشود.
سایر یافتههای فیزیکی که معمولاً با سندرم Singleton-Merten مرتبط است ممکن است شامل ضعف عضلانی عمومی بدن، از دست دادن تدریجی بافت عضلانی (atrophy)، اختلال در رشد که احتمالاً منجر به کوتاهی قد میشود، مشکلات بینایی (glaucoma)، رباطهای غیر طبیعی مفاصل و ماهیچه و یا ناهنجاری پا باشد. به نظر میرسد که در برخی موارد، سندرم سینگلتون-مرتن در نتیجه یک جهش تصادفی (sporadic) رخ میدهد. در موارد دیگر، الگوی توارث اتوزومال غالب پیشنهاد شده است.
علائم و نشانههای سندرم Singleton-Merten
سندرم Singleton-Merten، یک اختلال بسیار نادر، با ناهنجاریهای دندان (dental dysplasia)، تجمع غیرطبیعی رسوب کلسیم (calcifications) در شریان اصلی بدن (آئورت) و دریچههای خاص قلب (یعنی دریچههای آئورت و میترال) است که گاها با پوکی استخوان (osteoporosis) مشخص میشود.
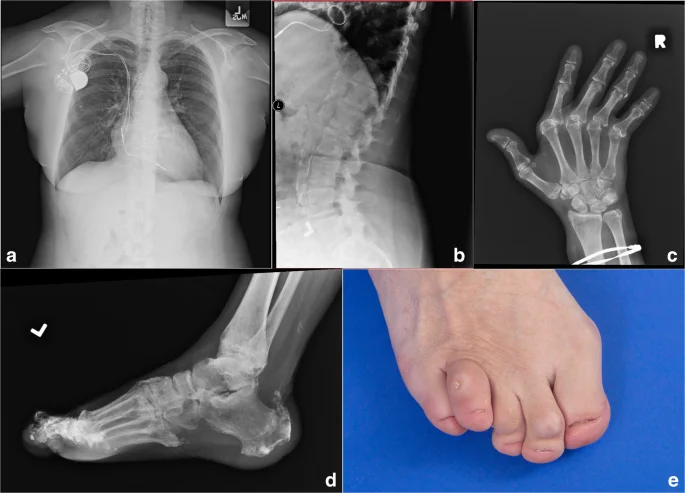
نوزادان مبتلا به سندرم Singleton-Merten بین سنین 4 تا 24 ماهگی، اکثرا ضعف عضلانی عمومی و از دست دادن یا تحلیل (atrophy) بافت عضلانی را تجربه میکنند. نوزادان مبتلا همچنین ممکن است در رشد جسمی (سوماتیک somatic) اختلال داشته باشند که احتمالاً منجر به کوتاهی قد و یا تاخیر در توانایی هماهنگ دادن عضلات و انجام فعالیتهای خاص میشود.
ناهنجاریهایی که روی دندانها تأثیر میگذارند نیز در سنین پایین در افراد مبتلا به سندرم سینگلتون-مرتن رخ میدهد. نوزادان مبتلا ممکن است دچار پوسیدگی (caries) شوند و به دنبال آن دندانهای شیری (deciduous) خود را زودتر از موعد از دست بدهند. برخی از دندانهای دائمی (erupt) ممکن است رشد نکنند یا ممکن است دیر رشد کنند و در صورتی که دندانهای دائمی رشد کنند معمولاً بدشکل هستند (dysplastic). در برخی بیماران، دندانهای دائمی نیز ممکن است زود از بین بروند.
در اواخر دوران نوزادی یا اوایل کودکی، افراد مبتلا ممکن است علائم مرتبط با تجمع تدریجی رسوبات کلسیم (calcifications) در شریان اصلی بدن (آئورت) و در دریچههای خاصی از قلب (یعنی آئورت و میترال) را تجربه کنند. آئورت از بطن چپ قلب سرچشمه میگیرد و خون غنی از اکسیژن را به تمام شریانهای بدن (به استثنای شریان ریوی) میرساند.
در افراد مبتلا به سندرم Singleton-Merten، رسوبات کلسیم در نزدیکترین قسمت آئورت به قلب (proximal thoracic aorta) تشکیل میشود. تجمع رسوبات کلسیم پیشرونده است و به طور معمول باعث انسداد و تنگ شدن آئورت (calcific aortic stenosis) میشود و جریان خون اکسیژن دار را مسدود میکند. در برخی موارد، رسوبات غیر طبیعی کلسیم نیز ممکن است در اطراف دریچه سمت چپ قلب ایجاد شود (کلسیفیکاسیون دریچه میترال).
در نتیجه کلسیفیکاسیون این ساختارهای مختلف، افراد مبتلا ممکن است فشار خون بالا (hypertension)، انقباضات غیر طبیعی قلب (systolic murmurs) و یا بزرگ شدن غیر طبیعی قلب (cardiomegaly) را تجربه کنند. در اواخر نوجوانی، قلب ممکن است نتواند خون را به طور موثر پمپاژ کند (نارسایی قلبی) که منجر به عوارض تهدید کننده زندگی میشود.
نوزادان مبتلا به سندرم Singleton-Merten همچنین ممکن است نازک شدن و ضعف غیر طبیعی استخوانها (پوکی استخوان) را تجربه کنند. در نتیجه، استخوانها اغلب شکننده هستند و ممکن است به راحتی بشکنند. پوکی استخوان ممکن است در جمجمه و استخوانهای بلند دستها و پاها رخ دهد، اما بیشتر در استخوانهای دست و انگشتان دیده میشود.
سایر یافتههای مرتبط با سندرم Singleton-Merten ممکن است شامل ناهنجاریهای لگن و پا باشد که ممکن است به دلیل ضعف عضلانی رخ دهد. برخی از افراد مبتلا ممکن است تجمع غیر طبیعی فشار مایع چشم (آب سیاه) و یا حساسیت غیر طبیعی به نور (photosensitivity) داشته باشند.
ویژگیهای معمولی صورت در افراد مبتلا به سندرم سینگلتون-مرتن از جمله خط موی بالا، پیشانی پهن، افتادگی پلکهای بالایی (ptosis)، از دست دادن شیار عمودی بین قاعده بینی و لب فوقانی (philtrum) و لبه بالایی نازک (vermillion) توصیف شده است.
علت ابتلا به سندرم Singleton-Merten
سندرم Singleton-Merten یک اختلال بسیار نادر است که با الگوی توراث اتوزمال غالب به ارث میرسد. افراد مبتلا به نوع خفیف سندرم سینگلتون-مرتن ممکن است تنها یک علامت خفیف از بیماری مانند بثورات پوستی (psoriasis) را نشان دهند ولی افراد با فرم شدید این سندرم ممکن است تمام ناهنجاریهای ساختار و اختالات اصلی سندرم Singleton-Merten، از جمله کلسیفیکاسیون (calcifications) در قلب و آئورت و همچنین ناهنجاریهای اسکلتی و دندانی را نشان دهند.
سندرم Singleton-Merten به دلیل جهش در ژن IFIH1 ایجاد میشود. این ژن مسئول ساخت پروتئینی به نام پروتئین مرتبط با تمایز ملانوما ژن 5 (MDA5- melanoma differentiation-associated gene 5 protein) است. هنگامی که این پروتئین به طور نرمال سنتز و فعالی میکند، MDA5 با حضور ویروسها فعال میشود.
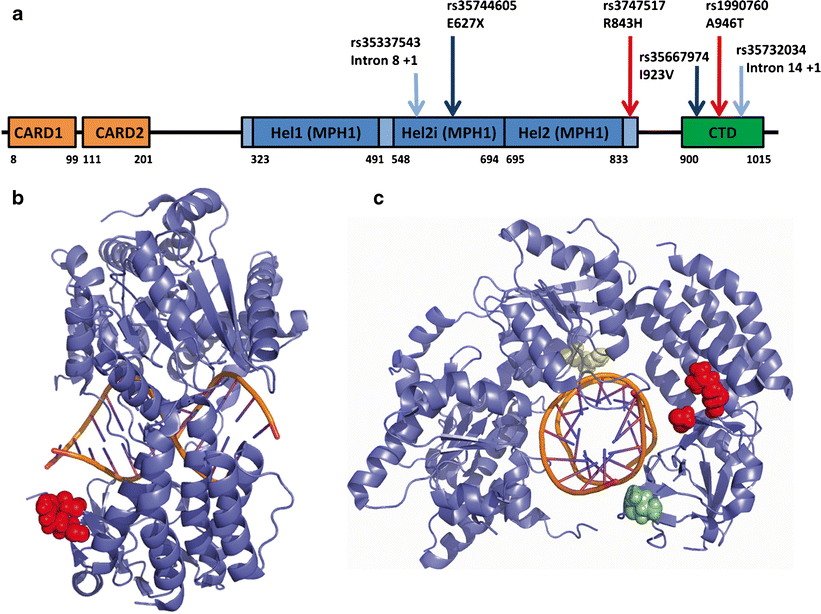
پس از فعال شدن منجر به تولید مولکولهای خاصی میشود که میتواند فعالیت سیستم ایمنی را افزایش دهد. با این حال، پروتئین MDA5 در افراد مبتلا به سندرم Singleton-Merten همیشه فعال است، حتی زمانی که هیچ ویروسی وجود نداشته باشد. این عمل منجر به یک سیستم ایمنی بیش فعال (overactive immune system) میشود. این حالت بیش فعال بودن سیستم ایمنی، ممکن است باعث اختلال در متابولیسم کلسیم در بدن شود که منجر به کلسیفیکاسیون غیرطبیعی آئورت و دریچههای قلب و از بین رفتن کلسیفیکاسیون در استخوانها میشود.
همچنین ژن دیگری به نام DDX58 شناسایی شده است که با سندرم آتیپیک Singleton-Merten مرتبط است. DDX58 ژنی است که مسئول ساخت پروتئینی به نام ژن القایی با اسید رتینوییک (RIG-I- retinoic-acid-inducible gene) است. یک خانواده از افراد مبتلا به این جهش تحت تاثیر مشکلات بینایی، کلسیفیکاسیون آئورت و ناهنجاریهای اسکلتی قرار گرفتند اما دارای دندان طبیعی بودند. خانواده دیگری از افراد مبتلا به این جهش مشکلات بینایی و ناهنجاریهای اسکلتی داشتند اما آئورت طبیعی و دندان طبیعی داشتند.
اختلالات با علائم مشابه
علائم بیماریهای زیر میتواند مشابه علائم سندرم سینگلتون – مرتن باشد. مقایسه و افتراق بین بین علائم این بیماریها میتواند برای تشخیص افتراقی مفید باشد:
- دیسپلازیهای اکتودرمی (ectodermal dysplasias – ED) گروهی از بیماریها هستند که معمولاً با ناهنجاریهای ناحیه سر و صورت (جمجمه)، مو، دندان، ناخن و یا پوست مشخص میشوند. در برخی موارد، ناهنجاریهای مرتبط با برخی از دیسپلازیهای اکتودرمی (ED) ممکن است مشابه یافتههای مرتبط با سندرم سینگلتون-مرتن باشد. برخی از دیسپلازیهای اکتودرمی ممکن است با ناهنجاریهای قلب و سیستم عروقی مشابه موارد مرتبط با سندرم Singleton-Merten مشخص شوند.
- سندرم Aicardi-Goutieres یک بیماری نادر است که مغز، سیستم ایمنی و پوست را تحت تاثیر قرار میدهد. این بیماری با شروع زودرس اختلال پیشرونده در عملکرد مغز مشخص میشود که معمولاً منجر به ناتوانی شدید ذهنی و جسمی میشود. افراد همچنین ممکن است بثورات پوستی خارش دار و دردناک (chilblains)، مشکلات بینایی (glaucoma)، از دست دادن دندان و بدشکلی پا داشته باشند. سندرم Aicardi-Goutieres با جهش در چندین ژن مختلف از جمله ژن IFIH1 که علت سندرم Singleton-Merten نیز است، همراه است.
تشخیص سندرم Singleton-Merten
تشخیص سندرم Singleton-Merten ممکن است در دوران شیرخوارگی بر اساس شناسایی علائم فیزیکی نوزاد (مانند ضعف و آتروفی عضلانی، ناهنجاریهای دندانی و تغییرات اسکلتی) قابل شناسایی باشند. تشخیص اولیه بیماری ممکن است با یک ارزیابی بالینی، شرح حال دقیق بیمار و انجام انواع آزمایشات تخصصی تایید شود. بررسی وجود رسوبات کلسیم در آئورت، در کنار سایر یافتههای شرح داده شده در بالا، مهر تاییدی بر تشخیص سندرم Singleton-Merten است.
آزمایشات اشعه ایکس (X ray) ممکن است برای تأیید وجود و میزان رسوبات کلسیم (calcifications) در آئورت و دریچههای قلبی استفاده شود. انسداد یا تنگی (stenosis) دریچههای قلب (به ویژه دریچههای آئورت و میترال) ممکن است با کاتتریزاسیون قلبی (cardiac catheterization) تایید شود.
در طی این روش، یک لوله توخالی کوچک (catheter) وارد یک سیاهرگ بزرگ میشود و از رگهای خونی منتهی به قلب عبور میکند. این روش به پزشکان اجازه میدهد تا میزان جریان خون در قلب را تعیین کرده و فشار داخل قلب را اندازه گیری کنند. مطالعات اشعه ایکس ممکن است برای تایید وجود و بررسی میزان پوکی استخوان نیز انجام شود. هنگامی که شکستگی استخوان بیشتر از حد معمول اتفاق میافتد، ممکن است میتوان به پوکی استخوان شک کرد.
آزمایشهای اشعه ایکس همچنین ممکن است منافذ توخالی بزرگ و غیر طبیعی قسمتهایی در استخوانها را که حاوی بافت چربی (حفرههای مغز استخوان) در استخوانهای دست و یا پا هستند (مانند metacarpals، carpals، phalanges)، نشان دهد.
درمانهای استاندارد
سندرم Singleton-Merten دارای علائم خاصی است که از طریق آن میتوان تا حدودی افراد مبتلا را تشخیص داد اما برای درمان این سندرم به تلاش منسجمِ تیمی از متخصصان نیاز است مانند متخصصان اطفال، جراحان ماهر، متخصصانی که ناهنجاریهای قلب را تشخیص داده و درمان میکنند (متخصصان قلب)، متخصصین دندانپزشکی، فیزیوتراپها و متخصصانی که مشکلات پوست را تشخیص داده و درمان میکنند (متخصصین پوست).
درمانهای خاص سندرم Singleton-Merten شامل درمانِ علائمی و حمایتی (symptomatic و supportive) هستند. منظور از درمانهای علامتی این است که تیم پزشکی تلاش میکند تا ناهنجاریهای حاصل از این بیماری، مانند مشکلات اسکلتی و یا اختلالات ایجاد شده در سلامت دهان و دندان این افراد را رفع کنند و منظور از درمان حمایتی در واقع مشاورههای مختلف روان شناسی و روان پزشکی به خانواده و خودِ فرد بیمار است.
خدمات ویژه ای که ممکن است برای کودکان آسیب دیده مفید باشد ممکن است شامل حمایتهای اجتماعی، فیزیوتراپی و سایر خدمات پزشکی، اجتماعی باشد. مشاوره ژنتیک برای افراد مبتلا و خانوادههای آنها مفید خواهد بود.
همچنین بخوانید:
- سندرم اهلرز-دانلوس (EDS)
- سندرم آپرت (Apert syndrome)
- آکندروپلازی چیست؟ علائم، علل، تشخیص و درمان
- سندرم امانوئل
مترجم: فاطمه فریادرس