


بیوانفورماتیک, تکنیک ها, ویدیو های آموزشی
هومولوژی مدلینگ (Homology Modeling) چیست؟

هومولوژی مدلینگ (Homology Modeling): پیش بینی و مدل سازی ساختار دوم پروتئین
پروتئین ها واحد های عملکردی سلول می باشند که از واحد های ساختاری اسیدهای آمینه که با پیوند پپتیدی به هم متصل شده اند، تشکیل شده اند. پروتئین ها ساختارهای متعددی دارند. قرار گرفتن اسیدهای نوکلئیک به ترتیب کنار هم ساختمان اول پروتئین (ساختار خطی) را تشکیل می دهند. به ساختاری که پروتئین در حین تاشدگی به خود می گیرد، ساختار دوم پروتئین گفته می شود که به دو دسته مارپیچ آلفا و صفحه های بتا تقسیم می شود.
در اثر کنار هم قرار گرفتن مارپیچ های آلفا و بتا توسط پیوندهای هیدروژنی، موتیف های پروتئینی تشکیل می شوند که در واقع موتیف به نوعی یک ساختار فوق ساختاری دوم می باشد. ترکیب چندین موتیف کنار هم تشکیل یک دمین پروتئینی را می دهد. به ساختمان سه بعدی پروتئین ها ساختمان سوم پروتئین گفته می شود. به چگونگی قرار گرفتن زنجیره های پروتئینی یا در واقع زیر واحد های پروتئینی کنار همدیگر، ساختمان چهارم پروتئین گفته می شود.
یک شکاف عمیق بین توالی های شناسایی شده پروتئین و ساختمان پروتئین وجود دارد، لذا روز به روز نیاز به درک صحیح ساختمان پروتئین ها احساس می شود. به همین دلیل محققان در زمینه ی بیوانفورماتیک حوزه ی پروتئین به دنبال ابزارهایی به منظور پیش بینی ساختار پروتئین می باشند که بتوانند به مهندسی پروتئین دست یابند. در اینجا به معرفی یکسری از پایگاه های داده به منظور پیشگویی ساختار دوم پروتئین می پردازیم.
پایگاه داده ی JPRED:
که به منظور پیشگویی ساختار دوم پروتئین به کار می رود. برای دسترسی به پایگاه داده ی JPRED از طریق سایت http://www.compbio.dundee.ac.uk/jpred اقدام می نماییم
پایگاه داده ی PSIPRED:

پایگاه داده ی PSIPREDیک پایگاه داده ی پرکاربرد دیگر به منظور پیشگویی ساختار دوم پروتئین می باشد که برای دسترسی به این پایگاه داده می توان از لینک http://bioinf.cs.ucl.ac.uk/psipred/ اقدام نمود.
پایگاه داده ی PredictProtein:
یک سایت مرجع پروتئین می باشد که ابزارهای متعددی برای مطالعات بیوانفورماتیک پروتئین دارد که برای دسترسی به این پایگاه داده به لینک https://www.predictprotein.org مراجعه نمایید.
GOR:
یک پایگاه داده ی کاربردی دیگر به منظور پیشگویی ساختار دوم پروتئین می باشد.
هومولوژی مدلینگ
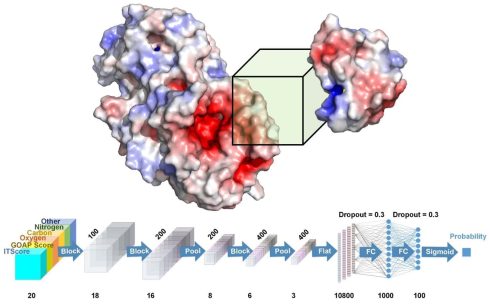
هومولوژی مدلینگ یک ابزار مفید برای پیش بینی ساختار پروتئین می باشد که تنها وقتی کارآمد است که داده های مربوط به توالی را داشته باشیم. اطلاعات ساختاری غالبا از اطلاعات مربوط به توالی با ارزش تر می باشد چون اطلاعات بیشتری در رابطه با عملکرد پروتئین در اختیار محقق می گذارد. برنامه های در دسترس برای هومولوژی مدلینگ از روش ها و رویکردهای مختلفی برای تولید مدل نهایی استفاده می کنند.
در هر کدام از مراحل مربوط به homology modelling، فاکتورهای مختلفی کیفیت مدل های تولید شده را تحت تاثیر قرار می دهد. همچنین انتخاب مناسب برنامه تاثیر بسزایی در بهبود کیفیت مدل نهایی دارد. نرم افزارهای مورد استفاده در هومولوژی مدلینگ به طور کلی به دو دسته ی نرم افزار های تحت وب و easy modeler تقسیم می شوند.
از جمله نرم افزار های تحت وب می توان به Swiss-MODEL، ESyPred3D، 3D-Jigsaw، HHPred و Phyre2 اشاره نمود. از جمله نرم افزار های easy Modeler می توان از BLAST، HHPred و Phyre2 برای بدست آوردن الگوهای مناسب استفاده نمود. همچنین می توان نرم افزار های Modeller و Phyton استفاده نمود.

نرم افزار MODELLER یک برنامه به منظور هومولوژی مدلینگ اتوماتیک پروتئین ها می باشد. نرم افزار MODELLER یکی از نرم افزارهای جامع به منظور همولوژی و یا مقایسه ی مدلینگ های ساختار سه بعدی پروتئین می باشد اما بسیاری از کاربران کار با MODELLER را کمی سخت می دانند. چرا که نیازمند command و دانش پایه ای پایتون می باشد تا بهترین کارایی را داشته باشد. به همین منظور EasyModeller که یک فاصل کاربری گرافیکی می باشد به جای MODELLER استفاده می شود.
داکینگ مولکولی

داکینگ مولکولی یا Molecular Docking از جمله ابزارهای بیوانفورماتیکی می باشد که کاربردهای وسیعی در علم بیوانفورماتیک بالاخص طراحی دارو دارد. به طور کلی اگر تعریفی در مورد داکینگ مولکولی داشته باشیم، می توان گفت که داکینگ مولکولی یک روش بیوانفورماتیکی و محاسباتی است که به دنبال اتصال مولکولی می باشد. در داکینگ مولکولی ما به دنبال حالت های اتصال لیگاند پروتئینی می باشیم.
از آنجایی که داکینگ مولکولی می تواند انواع اتصالات مولکولی را پیش بینی کند و در نتیجه قادر به پیش بینی اتصال لیگاندها به محل مناسبشان می باشد، کاربردهای وسیعی در علم پزشکی بخصوص طراحی دارو دارد. به عبارت ساده تر، بسیاری داکینگ مولکولی را مشابه چفت شدن قفل و کلید می دانند. به زبان دقیق و علمی، ما در داکینگ مولکولی بدنبال یک لیگاند می گردیم که از نظر انرژی و از نظر هندسی با مطابقت دقیقی با محل اتصال پروتئین داشته باشد.
نقش اساسی ارتباط بین پروتئین و سایر مولکول ها در فرآیندهای بیولوژیکی مهمی نظیر بیان ژن، رونویسی، انتقال سیگنال و … غیر قابل کتمان می باشد. از طرف دیگر بدست آوردن اطلاعات از نحوه ی اتصال پروتئین با سایر مولکول ها از طرق ازمایشگاهی نیازمند تکنیک های پیچیده و گران قیمتی نظیر کریستالوگرافی اشعه ایکس یا NMR می باشد، لذا تکنیک داکینگ مولکولی با استفاده از نرم افزارهای متعدد برای پیش بینی اتصال پروتئین با سایر مولکول ها یک تکنیک بیوانفورماتیک کارا محسوب می شود. که طبق آمار دقت تکنیک داکینگ مولکولی به طور متوسط 60 تا 75 درصد می باشد که عدد قابل قبولی است.
در مبحث داکینگ مولکولی می توان به انواع مختلف داکینگ مولکولی از جمله داکینگ پروتئین-لیگاند، داکینگ پروتئین-پروتئین، داکینگ پروتئین-پپتید، داکینگ آنتی بادی-آنتی ژن، داکینگ پروتئین-اسید نوکلئیک، داکینگ پروتئین-لیپید و داکینگ پروتئین-کربوهیدرات اشاره نمود. از آنجایی که در مبحث داکینگ به اتصال یک مولکول کوچک به نام لیگاند به یک پروتئین آنزیمی اشاره می کنیم، بهتر است بدانیم که این اتصال چه عواقبی در بیولوژی دارد و با دو اصطلاح آگونیسم و آنتاگونیسم آشنا شویم.
همانطور که اشاره شد در داکینگ مولکولی اتصال لیگاند به آنزیم می تواند منجر به فعال یا غیر فعال شدن آنزیم مورد نظر گردد که در صورت فعال شدن و ارسال یک پیام بیولوژیکی به این عمل آگونیسم و در صورت غیر فعال شدن به این عمل آنتاگونیسم گفته می شود.
مسلما داکینگ مولکولی نیز مانند سایر تکنیک های بیوانفورماتیکی یکسری محدودیت هایی دارد که اصلی ترین آن توانایی نمره گذاری توابع و عدم تاثیر گذاری یکسری واکنش های بین مولکولی مهم در نمره گذاری می باشد.
نرم افزارهای متعدد بسیاری در زمینه داکینگ مولکولی وجود دارد که برخی از این نرم افزارها تحت پشتیبانی سیستم عامل لینوکس و برخی از آنها تحت پشتیبانی سیستم عامل ویندوز می باشند. استفاده از سیستم عامل لینوکس شاید کمی برای افراد مبتدی در زمینه ی داکینگ مولکولی دشوار باشد بنابراین اگر کاربران به دنبال نرم افزارهایی هستند که تحت پشتیبانی ویندوز است، می توان به نرم افزارهای مهم و کاربردی Vina و Autodock اشاره نمود.
مسلما اگر نتایج داکینگ مولکولی با چندین نرم افزار چک شود صحت اطلاعات بیشتر بوده و از ایجاد پیش بینی های مثبت کاذب جلوگیری می شود. از جمله نرم افزارهای کاربردی در زمینه داکینگ مولکولی می توان به نرم افزار Vina، نرم افزار LeDock، نرم افزار MOE، نرم افزار Dock، نرم افزار rDock، نرم افزار Amber و نرم افزار Molegro اشاره نمود.
شبیه سازی دینامیک مولکولی
در حالی که مطالعات کریستالوگرافی به طور قانع کننده ای نقش مهم انعطاف پذیری پروتئین در اتصال لیگاند را نشان می دهد، هزینه و کار گسترده مورد نیاز برای تولید آنها باعث شده است که بسیاری به دنبال تکنیک های محاسباتی باشند که می توانند حرکات پروتئین را پیش بینی کنند. متأسفانه، محاسبات مورد نیاز برای توصیف حرکات کوانتومی-مکانیکی و واکنشهای شیمیایی سیستمهای مولکولی بزرگ اغلب برای بهترین ابررایانهها نیز بسیار پیچیده هستند.
شبیه سازی دینامیک مولکولی یا MD که برای اولین بار در اواخر دهه 1970 توسعه یافت، به دنبال غلبه بر این محدودیت با استفاده از تقریب های ساده مبتنی بر فیزیک نیوتنی برای شبیه سازی حرکات اتمی، در نتیجه کاهش پیچیدگی محاسباتی است. برای شبیه سازی دینامیک مولکولی در ابتدا یک مدل کامپیوتری از سیستم مولکولی از داده های رزونانس مغناطیسی هسته ای (NMR)، کریستالوگرافی، یا مدل سازی همسانی تهیه می شود، سپس نیروهای وارد بر هر یک از اتم های سیستم از طریق معادلاتی تخمین زده می شود.
به طور خلاصه، نیروهای ناشی از فعل و انفعالات بین اتم های پیوندی و غیر پیوندی نقش دارند. پیوندهای شیمیایی و زوایای اتمی با استفاده از فنرهای مجازی ساده مدلسازی میشوند و زوایای دو وجهی (یعنی چرخشهای حول یک پیوند) با استفاده از یک تابع سینوسی مدلسازی میشوند که تفاوتهای انرژی را تخمین میزند. نیروهای غیرپیوندی به دلیل برهمکنشهای واندروالس، مدلسازی شده با استفاده از پتانسیل لنارد-جونز، و برهمکنشهای باردار یا الکترواستاتیک، با استفاده از قانون کولمب، به وجود میآیند.
محدودیت های فعلی شبیه سازی دینامیک مولکولی:
جدای از این موفقیتها، کاربرد شبیهسازیهای دینامیک مولکولی هنوز توسط دو چالش اصلی محدود میشود. یکی میدانهای نیروی مورد استفاده نیاز به اصلاح بیشتری دارند، و دوم اینکه نیازهای محاسباتی بالا، شبیهسازیهای معمولی بیش از یک میکروثانیه را ممنوع میکند، که در بسیاری از موارد منجر به نمونه گیری ناکافی از سطح کانفورماسیون میشود.
به عنوان نمونه ای از این نیازهای محاسباتی بالا، در نظر بگیرید که یک شبیه سازی یک میکروثانیه از یک سیستم نسبتاً کوچک (تقریبا 25000 اتم) که روی 24 پردازنده اجرا می شود چندین ماه طول می کشد تا تکمیل شود. جدای از چالش های مربوط به نیازهای محاسباتی بالای این شبیه سازی ها میدان های نیروی مورد استفاده نیز تقریبی از مکانیک کوانتومی هستند در حالی که شبیهسازیها میتوانند بسیاری از حرکتهای مولکولی مهم را به دقت پیشبینی کنند، این شبیهسازیها برای سیستمهایی که اثرات کوانتومی برایشان مهم هستند، مناسب نیستند.
از کارآموزی های ژنیران دیدن فرمایید:
سلام، آیا امکان مدل سازی پروتئین هموتترامر توسط modeller وجود داره؟
سلام دوست عزیز اگر پروتئین در پایگاه های داده ها ثبت شده باشه بله امکانش هست
ممنون از پاسخ گوییتون، در کدوم پایگاه داده؟ یونی پرات یا پی دی بی؟ چون پروتئین من ساختار کریستالوگرافیکش در پی دی بی موجود نیست. با این وجود امکان مدل سازیش در مادلر وجود داره؟ اگر خیر برنامه های پیشنهادیتونچیه؟ ممنون از شما
مقاله عالیه دستتون درد نکنه