


دستهبندی نشده، ویکی ژن
RNA-seq: مبانی، کاربردها و پروتکل RNA-sequencing

RNA-seq چیست؟
RNA-seq (RNA-sequencing) تکنیکی است که می تواند کمیت و توالی RNA را در یک نمونه با استفاده از توالی یابی نسل بعدی (NGS) بررسی کند. رونوشت را تجزیه و تحلیل میکند و نشان میدهد که کدام یک از ژنهای کدگذاری شده در DNA ما، تا چه حد روشن یا خاموش هستند. در این مطلب به این نکته میپردازیم که چرا تکنیک RNA-Seq مفید است، این تکنیک چگونه کار میکند و پروتکل اساسی که امروزه معمولا استفاده میشود، چیست؟
کاربردهای RNA-sequencing چیست؟
RNA-seq به ما امکان می دهد تا رونوشت، کل محتوای سلولی RNA ها از جمله mRNA، rRNA و tRNA را بررسی و کشف کنیم. اگر بخواهیم اطلاعات موجود در ژنوم خود را با بیان پروتئین عملکردی آن مرتبط کنیم، درک رونویسی کلیدی است. RNA-seq میتواند به ما بگوید کدام ژنها در سلول روشن میشوند، سطح رونویسی آنها چقدر است و در چه زمانهایی فعال یا خاموش میشوند.
این به دانشمندان اجازه میدهد تا زیستشناسی یک سلول را عمیقتر درک کنند و تغییراتی را که در آن ایجاد میشود ارزیابی کنند. ممکن است نشان دهنده بیماری باشد برخی از محبوبترین تکنیکهایی که از RNA-seq استفاده میکنند عبارتند از: پروفایل رونویسی، شناسایی پلیمورفیسم تک نوکلئوتیدی (SNP)، ویرایش RNA و تجزیه و تحلیل بیان دیفرانسیل ژن.
RNA-seq می تواند اطلاعات حیاتی در مورد عملکرد ژن ها به محققان بدهد. به عنوان مثال، رونوشت میتواند تمام بافتهایی را که در آنها یک ژن با عملکرد ناشناخته روشن است، برجسته کند که ممکن است نقش آن را نشان دهد. همچنین اطلاعاتی را در مورد رویدادهای alternative splicing که رونوشت های مختلفی را از یک توالی ژن تولید می کند، جمع آوری می کند.
این رویدادها با توالی یابی DNA مشخص نمی شوند. همچنین میتواند تغییرات پس از رونویسی را که در طول پردازش mRNA رخ میدهد، مانند پلیآدنیلاسیون و 5’ cap را شناسایی کند. داده های RNA-seq از قرائت های کوتاه mRNA استفاده می کنند که عاری از DNA غیر کد کننده اینترونیک است، سپس این قرائت ها باید با ژنوم مرجع تراز شوند.
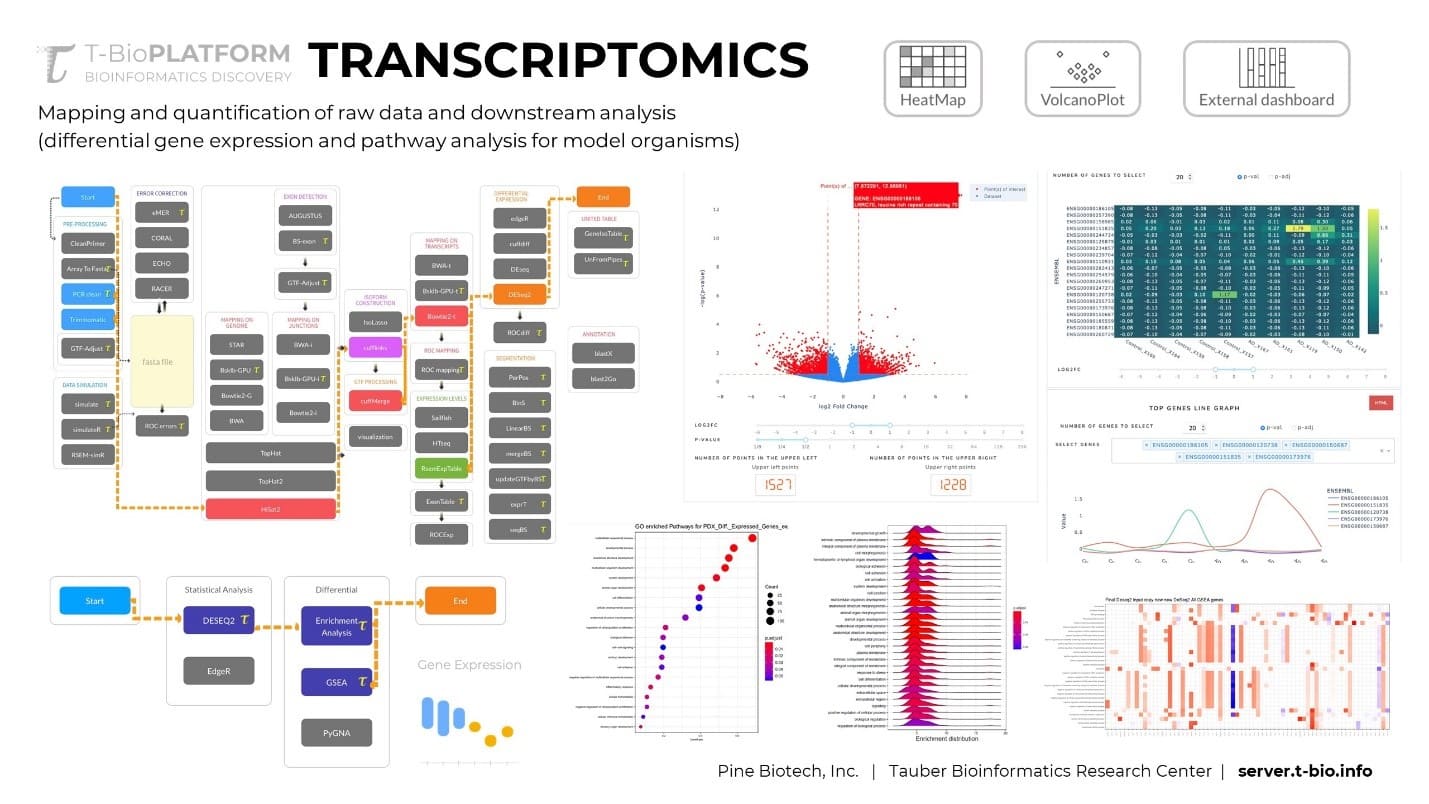
RNA-seq چگونه کار می کند؟
تکنیکهای اولیه RNA-seq از فناوری توالییابی سنگر استفاده میکردند، تکنیکی که اگرچه در آن زمان نوآورانه بود، با این وجود کم بازده و پرهزینه بود. اخیرا، با ظهور و گسترش فناوری NGS، ما توانستیم به طور کامل از پتانسیل RNA-seq استفاده کنیم.
یک گردش کار RNA-seq چندین مرحله دارد که به طور کلی می توان آنها را به صورت زیر خلاصه کرد:
- استخراج RNA(RNA extraction)
- رونویسی معکوس به cDNA(Reverse transcription into cDNA)
- اتصال آداپتور (Adapted ligation)
- تکثیر (Amplification)
- توالی یابی (Sequencing)
هنگامی که نمونه RNA خود را برای تجزیه و تحلیل به دست آوردید، اولین گام در این تکنیک شامل تبدیل جمعیت RNA برای تعیین توالی به قطعات DNA مکمل (cDNA) است. این کار با رونویسی معکوس انجام می شود و به RNA اجازه می دهد تا در یک گردش کار NGS قرار گیرد. سپس cDNA قطعه قطعه می شود و به هر انتهای قطعات آداپتورهایی اضافه می شود.
این آداپتورها حاوی عناصر عملکردی هستند که امکان توالی یابی را فراهم می کنند. پس از فرآیندهای تکثیر، انتخاب اندازه، پاکسازی و بررسی کیفیت، کتابخانه cDNA سپس توسط NGS تجزیه و تحلیل می شود و توالیهای کوتاهی را تولید میکند که مربوط به تمام یا بخشی از قطعهای است که از آن مشتق شده است.
عمقی (depth) که کتابخانه در آن توالی یابی می شود بسته به هدفی که داده های خروجی برای آن استفاده می شود متفاوت است. توالی یابی ممکن است از روش های توالی یابی single-end یا روش های توالی یابی paired-end پیروی کند.
توالی یابی single-end روشی ارزان و سریع است (تقریبا حدود یک درصد از هزینه توالی یابی سنگر) که قطعات cDNA را فقط از یک انتها توالی یابی می کند. روش های paired-end چون از هر دو طرف قطعات cDNA را توالی یابی می کنند، بنابراین گران تر هستند اما مزایایی را در بازسازی دادههای پس از توالی یابی ارائه میدهند.
باید بین پروتکل های strand-specific و non-strand-specific انتخاب صورت بگیرد. روش قبلی به این معنی است که اطلاعات مربوط به رشته DNA که رونویسی شده است حفظ می شود. ارزش اطلاعات اضافی به دست آمده از پروتکل های strand-specific آنها را به گزینه مطلوب تبدیل می کند.
این خوانش ها، که در پایان گردش کار، میلیونها مورد از آنها وجود خواهد داشت، در صورت موجود بودن میتوانند با ژنوم مرجع تراز شوند یا به صورت de novo مونتاژ شوند تا یک نقشه توالی RNA تولید کنند که رونوشت را پوشش میدهد.
چرا RNA-seq در مقایسه با تکنیک ریزآرایه (microarray) برتر در نظر گرفته می شود؟
تکنیک RNA-seq به طور گسترده ای برتر از سایر فناوری ها مانند هیبریداسیون ریزآرایه در نظر گرفته می شود. دلایل متعددی برای برتری تکنیک RNA-seq وجود دارد:
- محدود به توالیهای ژنومی نیست.
- برخلاف رویکردهای مبتنی بر هیبریداسیون، که ممکن است به پروب های گونهای خاص نیاز داشته باشد، تکنیک RNA-seq می تواند رونوشت هایی را از ارگانیسمهایی با توالیهای ژنومی که قبلا تعییننشده اند، شناسایی کند. این باعث می شود که اساسا برای تشخیص رونوشت های جدید، SNP ها یا سایر تغییرات برتری داشته باشد.
- سیگنال پسزمینه پایین: توالیهای cDNA مورد استفاده در RNA-seq را میتوان به مناطق هدف روی ژنوم map نمود، که حذف نویز آزمایش را آسان میکند. علاوه بر این، مسائل مربوط به هیبریداسیون متقابل (cross-hybridization) یا هیبریداسیون غیر استاندارد (sub-standard)، که میتواند آزمایشهای ریزآرایه را با مشکل مواجه کند، در آزمایشهای RNA-seq مشکلی ندارد.
- کمی تر هستند: داده های ریزآرایه فقط به صورت مقادیری نسبت به سایر سیگنال های شناسایی شده در ریزآرایه نمایش داده می شوند، در حالی که داده های RNA-seq قابل اندازه گیری هستند. تکنیک RNA-seq همچنین از مشکلات ریزآرایه ها در تشخیص سطوح رونویسی بسیار بالا یا بسیار پایین جلوگیری می کند.
آماده سازی کتابخانه cDNA
پس از در نظر گرفتن این نکات، می توانید شروع به تهیه کتابخانه cDNA خود کنید. این امر مستلزم قطعه قطعه شدن cDNA، افزودن توالی های آداپتوری، مخصوص پلتفرم و تکثیر cDNA است، اما روش دقیق برای پلتفرم مورد استفاده در این مرحله بسیار خاص خواهد بود. برای پروتکلهای strand-specific، تکثیر cDNA شامل سنتز رشته اول با واسطه رونویسی معکوس و سپس سنتز رشته دوم با واسطه DNA پلیمراز است.
همچنین ممکن است بارکدهایی اضافه شوند که multiplex را فعال می کنند، بنابراین نمونه های متعددی را می توان در یک run دستگاه توالی یابی کرد که می تواند به منظور کنترل کیفیت کتابخانه خود در پایان مرحله آماده سازی کتابخانه مفید باشد تا از موفقیت آمیز بودن پروتکل اطمینان حاصل نمایید.
توالی یابی cDNA
هنگامی که کتابخانه آماده شد، می توانید از پلتفرم توالی یابی انتخابی خود برای توالی کتابخانه cDNA خود به عمق یا depth و نیازهای دلخواه خود استفاده کنید. هنگامی که دادههای رونوشت شما تولید شد، میتوانید دادهها را به ژنوم مرجع خود map کنید یا اگر ژنوم مرجعی در دسترس نیست، آنها را به صورت de novo جمعآوری کنید.
فرآیند هم ترازی با وجود splice variant ها و modification ها، می تواند پیچیده تر شود و انتخاب ژنوم مرجع مورد استفاده نیز دشواری این مرحله را تغییر می دهد. بستههای نرمافزاری مانند STAR و ابزارهای کنترل کیفیت مانند Picard یا Qualimap در این مرحله مفید هستند. مونتاژ De novo امکان کشف رونوشتهای جدید را علاوه بر آنهایی که قبلا شناخته شدهاند، میدهد.
تجزیه و تحلیل داده های RNA-seq
پس از مرحله هم ترازی، می توانید روی تجزیه و تحلیل داده های خود تمرکز کنید. ابزارهایی مانند Sailfish، RSEM و BitSeq13 به شما کمک میکنند سطح رونویسی خود را کمی کنید، در حالی که ابزارهایی مانند MISO برای کمی کردن پیرایش متناوب ژن ها و تجزیه و تحلیل تخصصیتر در دسترس هستند. به طور خلاصه، RNA-seq امروزی به خوبی به عنوان گزینه برتر نسبت به ریزآرایه ها شناخته شده است و احتمالا در حال حاضر گزینه ارجح باقی خواهد ماند.
چالش های RNA-seq

در حدود یک دهه گذشته پیشرفت قابل توجهی در زمینه RNA-seq حاصل شده است. هزینه های مرتبط به طور قابل توجهی کاهش یافته است در حالی که توان عملیاتی افزایش یافته است. صحت توالی یابی به مراتب برتر از تکرارهای قبلی فناوری NGS است و در دسترس بودن ابزارهای تجزیه و تحلیل داده ها و pipeline ها به شدت بهبود یافته است.
با این حال، تعدادی از چالشها برای دانشمندان وجود دارد که باید هنگام بررسی آزمایشهای RNA-seq در نظر داشته باشند که شامل:
جداسازی RNA کافی و با کیفیت: در حالی که نیازهای کمیت نمونه برای آنالیز RNA-seq به شدت کاهش یافته است، هنوز مهم است که اطمینان حاصل شود که میتوانید RNA کافی برای برآورده کردن تمام نیازهای آنالیز خود، از جمله تکرار در صورت لزوم، به دست آورید.
همچنین مهم است که در نظر داشته باشید، در حالی که ممکن است RNA کل را جدا کنید، بسته به سؤال آزمایشی خود، احتمالا فقط کسری از این RNA ها (معمولا RNA پیام رسان (mRNA)) را توالی یابی می کنید که باعث کاهش بیشتر مقدار نمونه شما می شود.
این مقدار همچنین باید از کیفیت و خلوص بالایی برخوردار باشد زیرا نمونههای ضعیف احتمالا منجر به نتایج ضعیف یا در برخی موارد شکست در پروتکل آمادهسازی کتابخانه (library) میشوند. کیفیت و غلظت RNA را می توان با استفاده از طیف سنجی مرئی UV تعیین کرد. برخلاف DNA، RNA به سرعت تجزیه میشود، بنابراین مهم است که در تمام مراحل جداسازی و خالصسازی دقت کنیم.
تأثیر ادغام نمونه ها: ادغام نمونه ها قبل از آماده سازی کتابخانه (بدون استفاده از بارکد) می تواند تلاش و هزینه های توالی یابی را کاهش دهد یا در مواردی که تعداد نمونه بسیار محدود است، توالی یابی را فعال کند.
با این حال، در نظر گرفتن این موضوع در طول تجزیه و تحلیل دادهها، با توجه به اینکه یکی از این مخزنها به عنوان یک تکرار بیولوژیکی در نظر گرفته میشود، مهم است. تغییرات بین نمونه های تلفیقی می تواند منجر به نتایج گمراه کننده و مسائل آماری شود، بنابراین پیامدهای احتمالی باید در طول فرآیند طراحی آزمایش در نظر گرفته شود.
عمق توالی یابی در مقایسه با تعداد نمونه: ممکن است انجام هر چه بیشتر نمونه در یک توالی یابی واحد برای کاهش هزینه ها و زمان دستگاه، جذاب به نظر برسد. با این حال، این هزینه دارد. هر چه تعداد نمونه های مالتی پلکس بیشتر باشد، تعداد قرائت های کمتری برای هر یک از آن نمونه ها به دست می آید.
با کاهش عمق خواندن، عدم اطمینان در مورد قابلیت اطمینان دنباله های به دست آمده افزایش می یابد. فنآوریهای توالییابی هنوز تا کامل شدن فاصله دارند و اشتباهاتی در خواندن انجام میشود.
بنابراین مهم است که نقطه مناسب برای به دست آوردن عمق خواندن کافی برای اطمینان به کیفیت و صحت داده های توالی یابی به دست آمده و به حداکثر رساندن ظرفیت توالی یابی برای اطمینان از تجزیه و تحلیل تکرارهای بیولوژیکی کافی برای ارائه داده های معنی دار، پیدا شود.
مطالب مرتبط:
ممنون